新型α-葡萄糖苷酶抑制劑YG-18在小鼠體內的降糖作用、分子對接及急性毒性研究
2021-07-03 08:18:32閻成炟郭崇真張東虎趙凌霄林建陽中國醫科大學附屬第一醫院藥學部沈陽110001中國醫科大學藥學院沈陽110122
中南藥學 2021年6期
閻成炟,郭崇真,張東虎,趙凌霄,林建陽*(1.中國醫科大學附屬第一醫院藥學部,沈陽 110001;2.中國醫科大學藥學院,沈陽 110122)
糖尿病是一種以高血糖為主要臨床特點的常見的慢性內分泌性疾病。國際糖尿病聯盟的數據顯示,2015年全球糖尿病患病率為8.8%,預計2040年成人糖尿病患病率將上升到10.4%[1]。在我國成年人中,糖尿病的患病率高達11.6%[2],而其中占比非常高的便是2型糖尿病。2型糖尿病,即非胰島素依賴型糖尿病,是糖尿病最常見的類型,占所有糖尿病病例的90%以上[3]。控制餐后血糖是治療2型糖尿病的關鍵。
α-葡萄糖苷酶抑制劑可以抑制小腸刷狀緣上的α-葡萄糖苷酶對碳水化合物的轉化,從而延緩葡萄糖吸收,降低餐后血糖,在全球被廣泛應用于2型糖尿病的一線治療[4]。此外,除降糖活性,α-葡萄糖苷酶抑制劑還被發現與腫瘤轉移[5]、干擾宿主細胞的N-寡糖鏈生物合成以阻止病毒復制有關[6]。α-葡萄糖苷酶抑制劑具有起效快速且安全性高的特點。目前,在臨床上使用的上市藥物主要是阿卡波糖(acarbose)、伏格列波糖(voglibose)和米格列醇(miglitol)。傳統上對于α-葡萄糖苷酶抑制劑的開發與研究大多基于其擬糖骨架結構,例如亞胺糖、硫糖、二糖等結構[7-8]。然而,傳統的研究開發面臨著諸多問題,比如活性差、自然豐度低和立體化學過于復雜等[9]。因此,對于非糖α-葡萄糖苷酶抑制劑的開發具有廣闊前景[10-11]。
根據前期報道[12],將苯磺酰胺查爾酮結構整合到苯并吡喃中,設計得到的3-[4-(苯基磺酰胺)苯甲酰]-2H-1-苯并吡喃-2-酮結構,具有成為非糖α-葡萄糖苷酶抑制劑的巨大潛力。在前期研究中,該系列化合物中體外活性最強的化合物YG-18(如圖1)的半數抑制濃度(IC50)為(0.014±0.003)μmol·L-1,高于陽性對照藥物阿卡波糖。為了探究該藥物在體內的降低糖負荷后的血糖藥效和作用機制、藥物與酶活性部位可能的相互作用模式與安全性,本研究考察了該系列化合物中的YG-18在正常大鼠體內對蔗糖和葡萄糖負荷后的影響,并通過分子模擬分析研究了其對α-葡萄糖苷酶的抑制行為。最后,根據OECD急性毒性實驗指導原則[13]觀察了該藥物單次口服4 h、48 h和14 d的毒性反應、臟器情況和存活狀態的急性經口毒性。
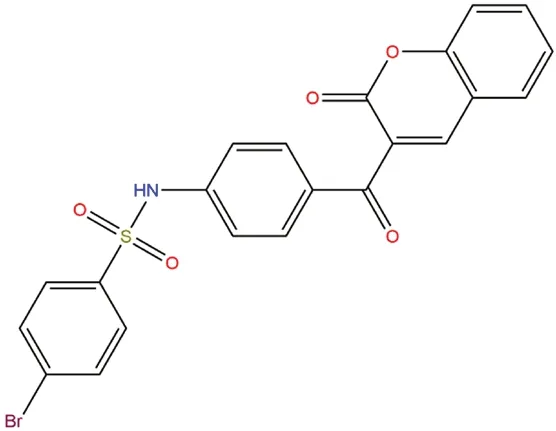
圖1 YG-18的化學結構Fig 1 Chemical structure of YG-18
1 材料
1.1 試藥
YG-18(沈陽藥科大學藥物化學教研室,純度≥ 98.0%);阿卡波糖(艾覽化工科技有限公司);二甲基亞砜(DMSO,天津市大茂化學試劑廠);聚氧乙烯蓖麻油EL、甘油(上海阿拉丁生化科技股份有限公司);純凈水(杭州娃哈哈集團有限公司);蔗糖、葡萄糖等試劑均為分析純。
1.2 儀器
One Touch Ultra Vue血糖儀[強生(中國)醫療器材有限公司];SQP電子天平(賽多利斯科學儀器有限公司);D3024R離心機(大龍興創實驗儀器股份公司);體重秤(慈溪市天東衡器廠);恒溫超聲儀(上海柯祁儀器設備有限公司)。
1.3 實驗動物
SPF級ICR小鼠(22~24 g),SPF級KM小鼠(22~24 g)[遼寧長生生物技術股份有限公司,動物生產許可證號:SCXK(遼)2020-0001]。
2 方法
2.1 YG-18單次給藥對正常ICR小鼠蔗糖、葡萄糖負荷后血糖影響
小鼠適應性喂養兩周后,ICR小鼠48只,隨機分為正常組、陽性藥物阿卡波糖組(Acar,9.6 mg·kg-1,ig)和YG-18組(7.23 mg·kg-1,ig)[14],每組8只,雌雄各半,每個實驗組平行設置兩組。其中藥物溶媒由0.67% DMSO、5% 聚氧乙烯蓖麻油EL、3% 甘油與生理鹽水組成。正常組給予空白溶媒進行灌胃給藥。阿卡波糖組、YG-18組灌胃對應的藥物。
禁食不禁水12 h后,平行設置的兩組小鼠分別灌胃給予蔗糖(4.0 g·kg-1)和葡萄糖(2.0 g·kg-1)負荷,用尾部采血法,通過血糖儀測定空腹血糖及給藥后0.5、1、2 h的血糖值,并計算血糖曲線下面積(AUC)。
2.2 分子對接實驗
利用Chemdraw 19.0繪制出阿卡波糖與YG-18的化學結構,使用Chem3D 19.0將其轉變為3D結構,并對其進行構象的優化。分子對接研究使用Discovery Studio v16.1進行。對接計算采用從PDB數據庫中獲得的α-葡萄糖苷酶與葡萄糖絡合物(PDB代碼:3A4A)的晶體結構。去除蛋白質結構中所有的結合水,并將所有的氫原子添進結構中。該蛋白質前期進行制備、優化和能量最小化處理。以α-葡萄糖苷酶共結晶的活性結合位點設置配體對接的空間,采用拉馬克遺傳算法進行對接實驗。對接的多種結合結果中,選取了能量最低的構象為本次實驗對藥物與酶抑制方式的預測構象。最后,使用Pymol和Discovery Studio v16.1繪制藥物與酶結合方式構象。
2.3 急性經口毒性實驗
根據OECD急性毒性實驗指導原則[13],將50只KM小鼠隨機分為4組給藥組與1組空白對照組,每組10只,雌雄各半。因YG-18無毒性資料可以參考且水溶性差,故給藥組采用的是OECD急性毒性實驗指導原則中推薦的175、550、1750、2000 mg·kg-1的序列給藥劑量,并使用羧甲基纖維素鈉配制混懸液灌胃給藥,除限定濃度2000 mg·kg-1外,其余對數劑量間隔1/2。
禁食不禁水12 h后,空白對照組小鼠給予溶媒羧甲基纖維素鈉單次灌胃處理,給藥組單次灌胃給藥處理,每組序貫進行。給藥當日連續觀察4 h,之后固定時間間隔觀察48 h。如果組內死亡小鼠數量大于半數,則降低下一組小鼠給藥劑量;如果組內死亡小鼠數量小于半數,則提高下一組小鼠給藥劑量。每一組存活動物均觀察14 d,后期死亡動物在統計時記為死亡。觀察結束后處死小鼠,解剖后觀察各組小鼠的器官的形態、顏色、質地等有無明顯變化。
2.4 統計學方法
使用SPSS 25.0統計軟件進行統計學分析,數據采用均數±標準差(±s)表示,分析模型選取單因素方差分析(one-way ANOVA),P<0.05表示差異有統計學意義,實驗中的數據采用Graph Pad Prism 8繪制作圖。
3 結果
3.1 YG-18單次給藥對正常ICR小鼠口服蔗糖、葡萄糖負荷后血糖水平影響
為了驗證YG-18在體外降低餐后血糖的藥效和作用機制,選取正常ICR小鼠作為模型,單次給藥后,分別記錄蔗糖、葡萄糖負荷后的血糖水平的血糖值變化,觀察其對血糖的影響,結果見圖2。
YG-18在7.23 mg·kg-1的劑量下單次給藥能降低正常小鼠蔗糖負荷后血糖的峰值(P<0.05),且YG-18組的血糖濃度-時間曲線下面積顯著降低(P<0.001)(見圖2A)。而在圖2B中,YG-18在7.23 mg·kg-1劑量下單次給藥對正常小鼠葡萄糖負荷后的各時間點的血糖和血糖濃度-時間曲線下面積均無明顯影響(P>0.05)。
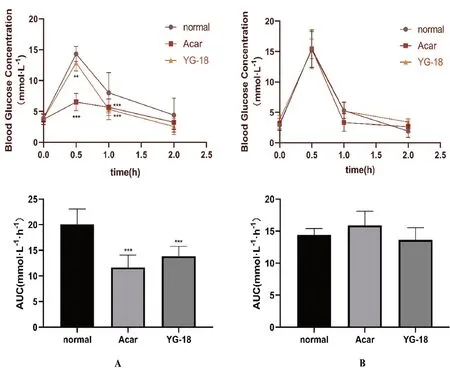
圖2 YG-18(7.23 mg·kg-1)對正常ICR小鼠血糖及血糖濃度-時間曲線下面積的影響Fig 2 Effect of YG-18(7.23 mg·kg-1)on the blood glucose and the blood glucose concentration time curve in ICR rats
由此可以證明,新型化合物YG-18的體內降糖作用的靶點為α-葡萄糖苷酶,只能對非單糖轉化為葡萄糖產生抑制作用,進而發揮降糖作用,而對直接攝入葡萄糖的血糖變化無法發揮作用。
3.2 分子對接
本次實驗分別采用了3D與2D方式展示了藥物-酶的結合構象,見圖3、4。圖3展示了α-葡萄糖苷酶-阿卡波糖之間的相互結合的3D與2D構象,圖4展示了α-葡萄糖苷酶-YG-18相互結合的3D與2D構象。結果顯示,阿卡波糖與酶結構中GLN 279、GLU 411和ARG442等氨基酸殘基形成氫鍵,氫鍵也是其與酶結合的主要方式。YG-18可能會與酶結構中的GLN 279、ARG 315、GLU 411、ASN 415和ARG 442氨基酸殘基形成氫鍵;除氫鍵外,其可能還與ASP 69、ASP 215、GLU 277氨基酸殘基形成范德華力。YG-18展現出的α-葡萄糖苷酶抑制活性可能與這樣的結合方式有關,且兩種藥物共同結合的ASP 69、GLU 277、GLN 279、GLU 411、ARG 442等氨基酸殘基可能為抑制活性的關鍵氨基酸。
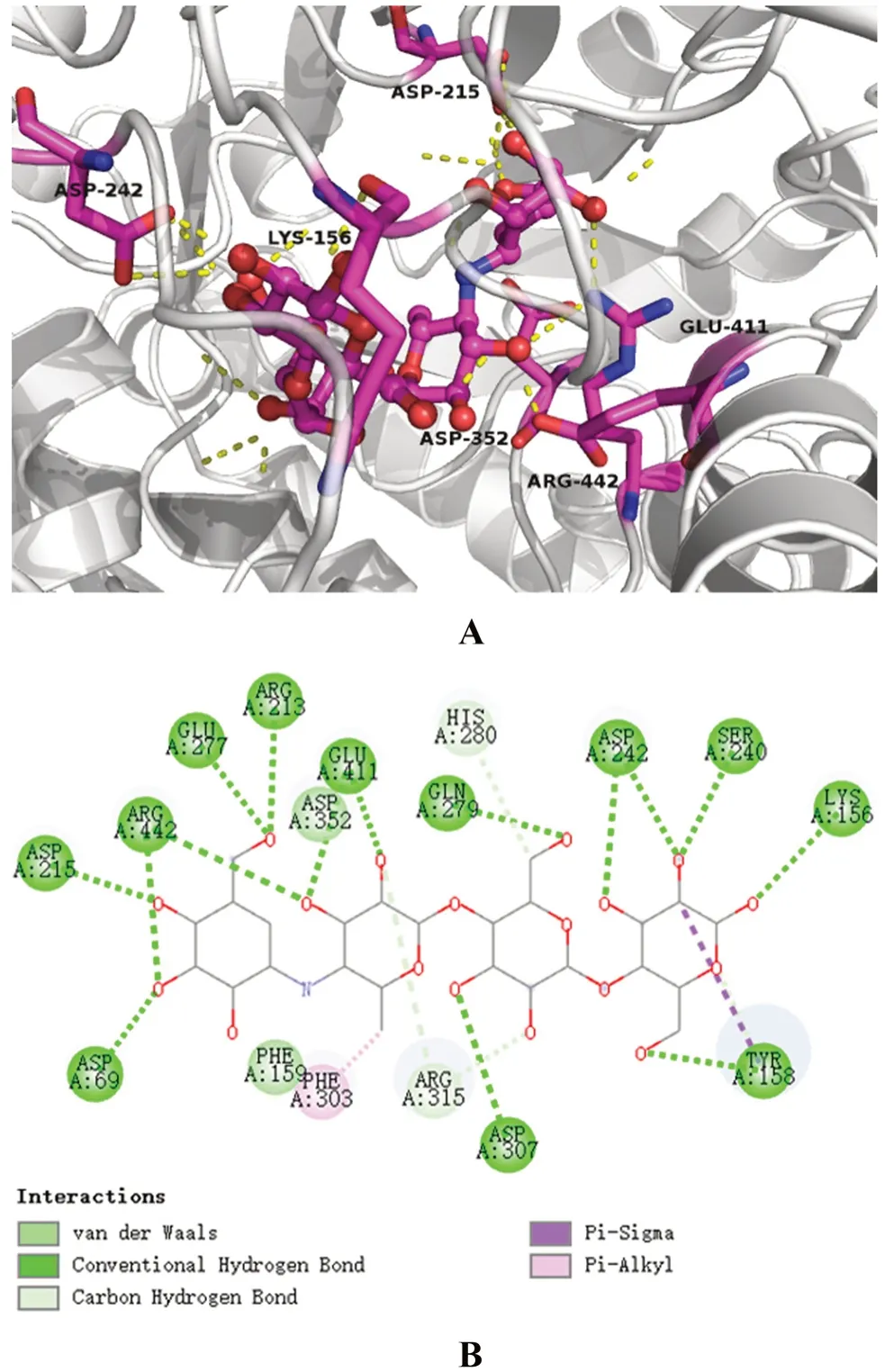
圖3 阿卡波糖-α-葡萄糖苷酶結合體的3D(A)和2D(B)結構Fig 3 3D(A)and 2D(B)structure of acarbose-α-glucosidase
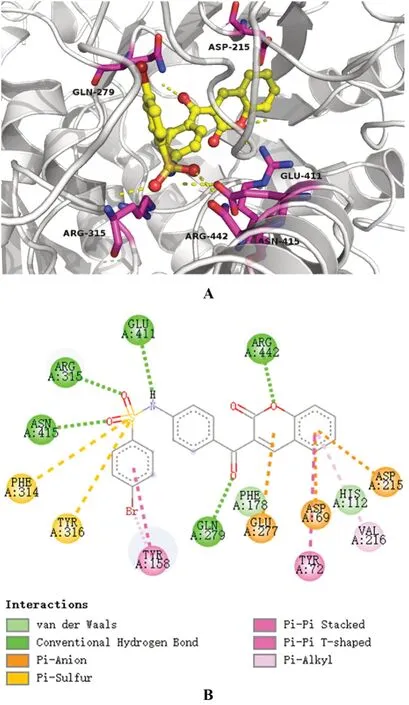
圖4 YG-18-α-葡萄糖苷酶結合體的3D(A)和2D(B)結構Fig 4 3D(A)and 2D(B)structure of YG-18-α-glucosidase
3.3 急性經口毒性實驗
OECD指導原則下YG-18單次給藥的序列劑量中,未能得出該藥物的LD50。各給藥組給藥后4 h內,小鼠開始出現不良反應,主要表現為腹瀉,而腹瀉為α-葡萄糖苷酶的主要不良反應之一。腹瀉在給藥后12 h內全部恢復。除腹瀉外,各劑量組均無其他不良反應,未出現死亡,根據全球統一毒性分級系統(Globally Harmonized Classification System)分類,YG-18可以被認為是幾乎無毒的。
在實驗結束后對所有小鼠進行頸椎脫臼處死,解剖后觀察,發現各組小鼠除有2只存在腸道梗阻外,其余小鼠器官的形態、顏色、質地都未發現顯著異常。根據小鼠發生腹瀉的毒性反應推測,該藥物的毒性反應主要是通過α-葡萄糖苷酶導致碳水化合物吸收緩慢,從而導致腹瀉。
4 討論
在本次研究中,化合物YG-18降低了蔗糖負荷后正常ICR小鼠的血糖峰值,并明顯降低了負荷2 h內的血糖濃度-時間曲線下面積。由此可見,YG-18雖然對于血糖峰值的影響不如陽性對照藥阿卡波糖,但是在1 h時的血糖可以控制在與阿卡波糖相同的水平,且2 h內的血糖濃度-時間曲線下面積顯著下降。在葡萄糖負荷后,阿卡波糖和YG-18組的血糖均未下降。根據α-葡萄糖苷酶抑制劑的作用特點,這證明YG-18是通過抑制小腸刷狀緣上的α-葡萄糖苷酶起到降血糖作用的。
本研究利用α-葡萄糖苷酶共結晶蛋白模型分析了YG-18與α-葡萄糖苷酶相互作用的潛在機制,發現YG-18主要通過氫鍵、范德華力、π-陰/陽離子、π-π相互作用與α-葡萄糖苷酶結合,而阿卡波糖中主要的相互作用只有氫鍵和范德華力。兩種藥物通過不同結合作用共同抑制的氨基酸殘基主要有ASP 69、GLU 277、GLN 279、GLU 411、ARG 442等,說明這些氨基酸殘基可能是藥物對α-葡萄糖苷酶產生抑制作用的關鍵氨基酸。
在急性經口毒實驗中,雖然本次實驗未能得出該YG-18的LD50值,但是在2000 mg·kg-1,小鼠在14 d內均未出現死亡及其他毒性反應,各臟器也未出現器質性改變,說明YG-18在2000 mg·kg-1劑量內具有很好的安全性,LD50>2000 mg·kg-1。
在合成工藝方面看來,目前,已上市的α-葡萄糖苷酶抑制劑阿卡波糖的工業合成途徑主要為游動放線菌(Actinoplanes spp.)發酵生產[15],由于是微生物途徑合成生產,在合成過程中需對菌株選育、發酵條件等條件嚴格控制,合成工藝非常復雜。與生物合成工藝相比,通過有機化學合成的新型α-葡萄糖苷酶抑制劑YG-18的工藝則較為簡單,且后續路線優化過程簡便快捷,可以大規模合成,合成純度也可以保證。所以就合成工藝而言,通過有機化學合成的新型α-葡萄糖苷酶抑制劑具有很大的優勢。
有研究表明,α-葡萄糖苷酶抑制劑不僅有降糖作用,還可以通過抑制腫瘤細胞表面N-糖基化蛋白的表達抗癌細胞遷移作用[16];此外,α-葡萄糖苷酶抑制劑還可以通過抑制宿主細胞中參與順序修建病毒包膜糖蛋白N-連接寡糖的α-葡萄糖苷酶從而抑制病毒的復制,達到抗病毒作用,現在已被證明可以被其抑制的病毒有登革病毒(DENV),黃熱病病毒(YFV)和寨卡病毒(ZIKV)等[17-19]。現有已上市的α-葡萄糖苷酶抑制劑吸收均較差,只在腸道內產生局部作用。然而無論是抗腫瘤還是抗病毒作用,僅依靠腸道內的局部作用顯然是無法實現的,如新型α-葡萄糖苷酶抑制劑具有較高的生物利用度,可為后續開發研究其體內抗腫瘤、抗病毒作用提供支持。
綜上所述,本研究為YG-18的降糖作用、潛在抑制機制和安全性提供了一定的參考和依據,同時也為后續對于3-[4-(苯基磺酰胺)苯甲酰]-2H-1-苯并吡喃-2-酮母核結構的化合物開發更加強效的α-葡萄糖苷酶抑制劑提供了依據。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
保健醫苑(2022年6期)2022-07-08 01:26:34
家庭科學·新健康(2022年3期)2022-05-10 00:32:13
學苑創造·A版(2020年9期)2020-10-13 09:41:02
家庭醫學(下半月)(2020年1期)2020-05-11 02:05:44
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
媽媽寶寶(2017年3期)2017-02-21 01:22:30
飼料與畜牧(規模養豬)(2016年5期)2016-12-01 03:48:40
人人健康(2016年13期)2016-07-22 10:34:06
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
