基于巰基化衍生的氣相色譜-質(zhì)譜法測(cè)定有機(jī)相及水相中氯化氰
2021-07-01 04:10:34李曉森吳姬娜夏俊美
色譜 2021年8期
李曉森, 吳姬娜, 夏俊美, 袁 鈴*, 楊 旸*
(1. 國民核生化災(zāi)害防護(hù)國家重點(diǎn)實(shí)驗(yàn)室, 北京 102205; 2. 防化研究院分析化學(xué)實(shí)驗(yàn)室, 北京 102205)
氰化物是一種常用的工業(yè)原料,因其對(duì)人體的高毒性和對(duì)環(huán)境的高風(fēng)險(xiǎn)性,而成為毒劑分析領(lǐng)域的研究熱點(diǎn)[1-4]。氯化氰(ClCN)是一種能夠引起全身中毒的劇毒氣體,屬于《禁止化學(xué)武器公約》附表中頒布的化學(xué)戰(zhàn)劑之一[5]。我國2001年即建立了水中ClCN的標(biāo)準(zhǔn)檢測(cè)方法,并制定了相應(yīng)的含量標(biāo)準(zhǔn),要求自來水中ClCN含量必須≤70 μg/L[6]。水中ClCN的分析主要采用分光光度法,檢測(cè)機(jī)理為氰根與巴比妥酸進(jìn)行顯色反應(yīng),然后測(cè)定反應(yīng)物的吸收波長[7-10]。該方法的缺陷在于分析穩(wěn)定性較差,采樣和測(cè)定必須在1 d內(nèi)完成[11],且無法實(shí)現(xiàn)ClCN的直接測(cè)量分析。
目前國內(nèi)外已有采用氣相色譜法(GC)檢測(cè)ClCN的研究[12-16],但是GC僅依靠保留時(shí)間實(shí)現(xiàn)化合物的區(qū)分,在樣本雜質(zhì)干擾較大或目標(biāo)物標(biāo)準(zhǔn)品難以獲得的情況下,無法對(duì)目標(biāo)物進(jìn)行準(zhǔn)確定性及定量。采用色譜-質(zhì)譜技術(shù),通過分析目標(biāo)物特定的保留時(shí)間及質(zhì)譜碎片,能夠?qū)崿F(xiàn)化合物的準(zhǔn)確定性分析及定量檢測(cè)。但ClCN的相對(duì)分子質(zhì)量小,沸點(diǎn)低(12.6 ℃),且極性較強(qiáng),當(dāng)采用GC-MS分析時(shí),ClCN的色譜出峰情況較差,檢測(cè)靈敏度低[17,18]。Nagashima等[19]使用便攜式氣相色譜-質(zhì)譜儀對(duì)ClCN直接分析,檢出限為10 mg/m3。Yang等[20]嘗試對(duì)ClCN直接進(jìn)行GC-MS分析,檢出限高達(dá)1.2~1.7 mg/L,無法滿足痕量檢測(cè)的要求。同時(shí),ClCN在常溫下?lián)]發(fā)性高且毒性大,因此直接對(duì)樣品中未知濃度的ClCN進(jìn)行分析具有一定安全隱患。采用液相色譜-質(zhì)譜法(LC-MS)對(duì)ClCN進(jìn)行分析時(shí),ClCN會(huì)在液相色譜的流動(dòng)相中發(fā)生水解,影響定量分析結(jié)果[21]。
通過衍生化手段將低沸點(diǎn)、極性強(qiáng)且色譜分離效果差的氯化氰衍生為極性較弱且穩(wěn)定性高的衍生化產(chǎn)物,進(jìn)一步通過GC-MS對(duì)氯化氰的衍生產(chǎn)物進(jìn)行分析,能夠獲得良好的色譜分離效果以及高檢測(cè)靈敏度,滿足氯化氰的痕量檢測(cè)要求。目前針對(duì)ClCN衍生方法的相關(guān)研究報(bào)道較少。巰基化衍生是衍生試劑分子中活潑的巰基(-SH)與目標(biāo)物直接進(jìn)行反應(yīng)的過程。已有研究報(bào)道[22]將巰基化衍生技術(shù)應(yīng)用于化學(xué)毒劑(如糜爛性毒劑路易氏劑)等的分析中。與傳統(tǒng)的硅烷化衍生[23]等柱前衍生方法相比,巰基化衍生過程通常無需加熱,僅在室溫下反應(yīng)數(shù)分鐘即可完成,大大縮短了衍生時(shí)間。本研究通過巰基化衍生的方法,采用1-丁基硫醇作為衍生化試劑,將ClCN衍生為硫氰酸酯,并進(jìn)行氣相色譜-質(zhì)譜的定性分析及定量檢測(cè)。以異丙基二硫醚作為內(nèi)標(biāo)物,采用內(nèi)標(biāo)法對(duì)有機(jī)相中的ClCN衍生產(chǎn)物進(jìn)行分析;應(yīng)用頂空-固相微萃取(HS-SPME)技術(shù)對(duì)水中ClCN的巰基化衍生產(chǎn)物進(jìn)行富集,并采用外標(biāo)法對(duì)水相中的ClCN衍生產(chǎn)物進(jìn)行分析。本研究建立的樣品制備及分析方法靈敏度高,選擇性好,樣品分析結(jié)果穩(wěn)定,適合用于環(huán)境中ClCN的分析。
1 實(shí)驗(yàn)部分
1.1 儀器與試劑
7890A/5975C氣相色譜-質(zhì)譜聯(lián)用儀(美國Agilent公司); Trace GC Ultra/TSQ Quantum XLS氣相色譜-串聯(lián)質(zhì)譜儀(美國Thermo公司)。
甲苯、二氯甲烷、氯仿購自北京百靈威科技有限公司(色譜純,純度≥99%);乙酸、氯化鈉、硫酸鈉、硼酸鈉、氫氧化鈉購自國藥集團(tuán)化學(xué)試劑北京有限公司(分析純,純度≥97%);去離子水購自屈臣氏;1-丁基硫醇、1-丙基硫醇、1-乙基硫醇、3,4-二巰基甲苯、異丙基二硫醚、三乙胺購自美國Sigma公司(分析純,純度≥97%)。聚二甲基硅氧烷(PDMS)固相微萃取纖維購自美國Supelco公司(膜厚度100 μm)。ClCN由實(shí)驗(yàn)室按照文獻(xiàn)[24]路徑合成,純度≥99%。河水采自北京市昌平區(qū)陽坊鎮(zhèn)京密引水渠;自來水由北京市供應(yīng);飲用水為娃哈哈瓶裝礦泉水。
1.2 樣品制備與衍生條件
準(zhǔn)確稱取兩份2.0 mg ClCN樣品,置于4.0 mL螺紋玻璃瓶中,各加入2.0 mL甲苯及去離子水,配制1.0 mg/mL ClCN/甲苯和ClCN/去離子水溶液,并充分振蕩溶解。
移取500 μL ClCN/甲苯和ClCN/去離子水溶液,置于1.5 mL螺紋玻璃瓶中,分別加入5 μL 1-丁基硫醇。根據(jù)反應(yīng)體系的不同,加入5 μL三乙胺(甲苯溶液)或5 μL 1.0 mmol/L氫氧化鈉溶液(水溶液)調(diào)節(jié)反應(yīng)體系的pH值至9,將螺紋玻璃瓶蓋緊后混合均勻,于40 ℃加熱條件下衍生反應(yīng)10 min,反應(yīng)結(jié)束后自然冷卻至室溫。
衍生反應(yīng)方程式如下:

CH3CH2CH2CH2S-CN+HCl
(1)
1.3 樣品前處理
衍生完畢后,在ClCN/甲苯衍生樣品中加入5 μL含10 mg/L異丙基二硫醚的甲苯溶液,振蕩混合均勻后取1.0 μL進(jìn)行GC-MS分析。
對(duì)于ClCN/去離子水衍生樣品,衍生完畢后,采用固相微萃取的方式進(jìn)行樣品前處理。將PDMS固相微萃取纖維通過瓶蓋隔墊插入螺紋玻璃瓶中,萃取纖維距離液面約5 mm。衍生反應(yīng)體系在55 ℃加熱條件下完成頂空-固相微萃取,萃取時(shí)間5 min。萃取完畢后拔出固相微萃取纖維,直接插入氣相色譜-質(zhì)譜聯(lián)用儀的進(jìn)樣口脫附2 min。
1.4 儀器條件
1.4.1色譜條件
色譜柱:Agilent DB-5MS柱(30 m×0.25 mm×0.25 μm);載氣:氦氣;恒流模式,流速:1.0 mL/min;進(jìn)樣口溫度:250 ℃;不分流進(jìn)樣:0.7 min;溶劑延遲:3 min;柱升溫程序:起始溫度40 ℃,保持1 min,以10 ℃/min的升溫速率升至280 ℃,保持5 min。
1.4.2質(zhì)譜條件
離子源EI源;離子源溫度250 ℃;四極桿溫度230 ℃;傳輸線溫度250 ℃;電子能量70 eV。全掃描分析時(shí),掃描范圍及速率:m/z29~550,0.5 s;子離子掃描時(shí),分別選擇m/z115(碰撞能量10 eV)及m/z57(碰撞能量5、10及20 eV)作為母離子;選擇離子監(jiān)測(cè)模式(SIM)時(shí),ClCN衍生產(chǎn)物定性和定量離子的m/z分別為115和41,內(nèi)標(biāo)物定性和定量離子的m/z分別為150和43。
1.5 安全措施
所有涉及ClCN的操作均需要佩戴相應(yīng)的防護(hù)器材,并在通風(fēng)良好的狀況下進(jìn)行。配制飽和的氫氧化鈉水溶液作為洗消劑,各種直接接觸ClCN的實(shí)驗(yàn)器材在使用后立即洗消(操作人員經(jīng)過專業(yè)培訓(xùn),配備相應(yīng)急救措施)。
2 結(jié)果與討論
本研究首先考察直接進(jìn)樣條件下氯化氰的色譜行為及質(zhì)譜出峰情況,接著對(duì)ClCN進(jìn)行巰基化衍生,并進(jìn)行正交試驗(yàn),以獲得最優(yōu)反應(yīng)條件。通過GC-MS/MS對(duì)ClCN的衍生產(chǎn)物進(jìn)行質(zhì)譜裂解規(guī)律解析,最終建立GC-MS/SIM定量分析方法并進(jìn)行方法學(xué)考察。
2.1 直接分析ClCN的色譜行為考察
取1 μL 10 mg/LClCN/甲苯標(biāo)準(zhǔn)工作溶液,按1.3節(jié)所述進(jìn)行全掃描分析。如圖1所示,目標(biāo)物的出峰時(shí)間非常靠前(tR為1.62 min)且色譜峰出現(xiàn)嚴(yán)重拖尾,難以對(duì)ClCN進(jìn)行準(zhǔn)確定量。這可能是因?yàn)镃lCN相對(duì)分子質(zhì)量較小,沸點(diǎn)低且極性強(qiáng),這也是目前對(duì)ClCN進(jìn)行色譜分析的難點(diǎn)所在。因此本研究采用衍生的方式,將ClCN衍生為沸點(diǎn)較高、極性較弱的衍生產(chǎn)物,以獲得更好的色譜峰形,并實(shí)現(xiàn)ClCN的定量分析。
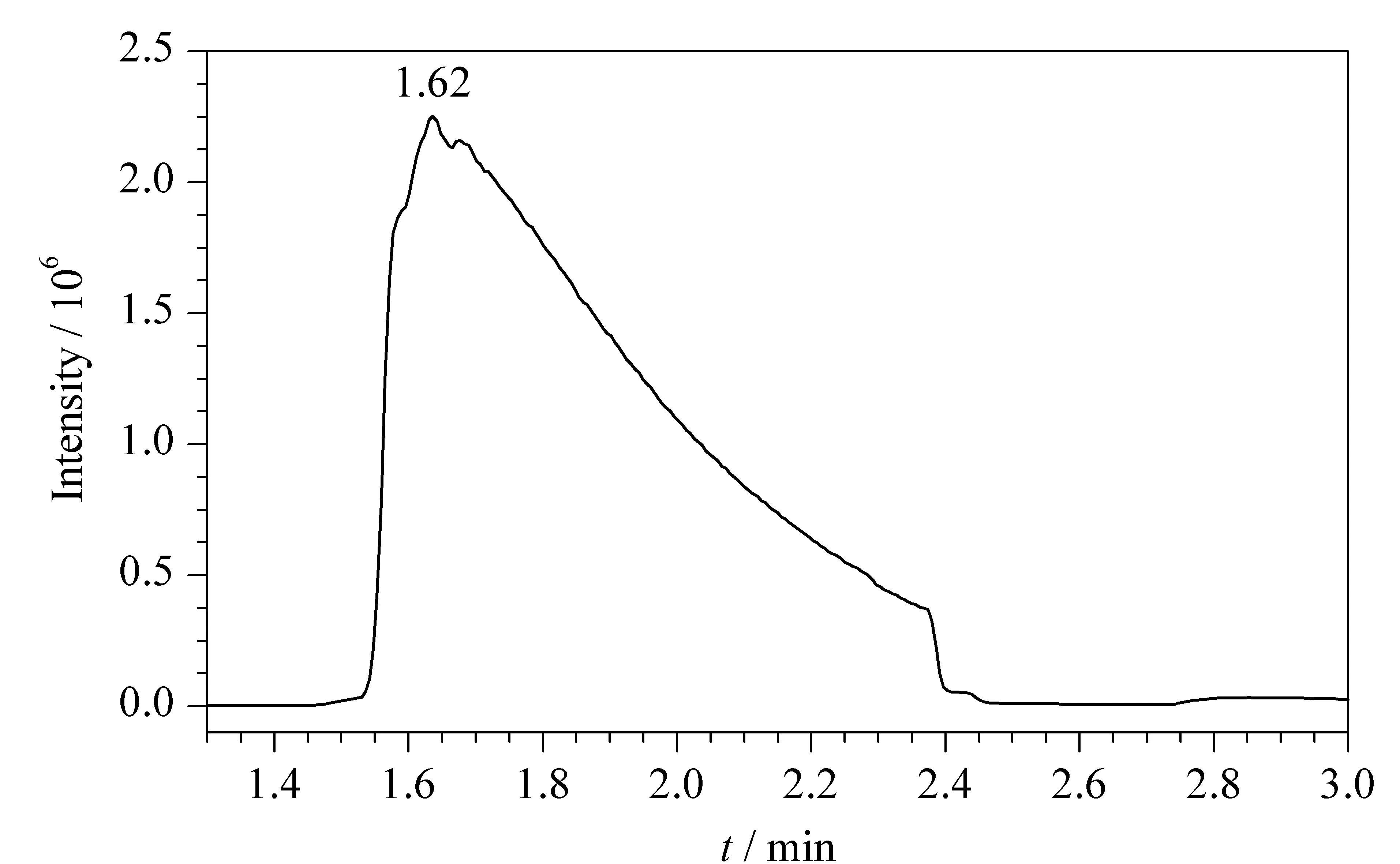
圖 1 ClCN(10 mg/L)/甲苯標(biāo)準(zhǔn)工作溶液的色譜圖Fig. 1 Chromatograms of ClCN (10 mg/L)/toluene standard working solutionCICN: cyanogen chloride.
2.2 衍生試劑的選擇
以甲苯為溶劑分別考察了有機(jī)相中4種巰基化衍生試劑(1-乙基硫醇、1-丙基硫醇、1-丁基硫醇和3,4-二巰基甲苯)的衍生效果,實(shí)驗(yàn)結(jié)果如圖2所示。采用1-乙基硫醇和3,4-二巰基甲苯進(jìn)行衍生后找不到衍生產(chǎn)物的色譜峰。采用1-丁基硫醇衍生后,ClCN衍生產(chǎn)物的峰面積大于1-丙基硫醇。因此本實(shí)驗(yàn)采用1-丁基硫醇作為巰基衍生試劑,衍生產(chǎn)物為硫氰酸丁酯,保留時(shí)間為7.468 min
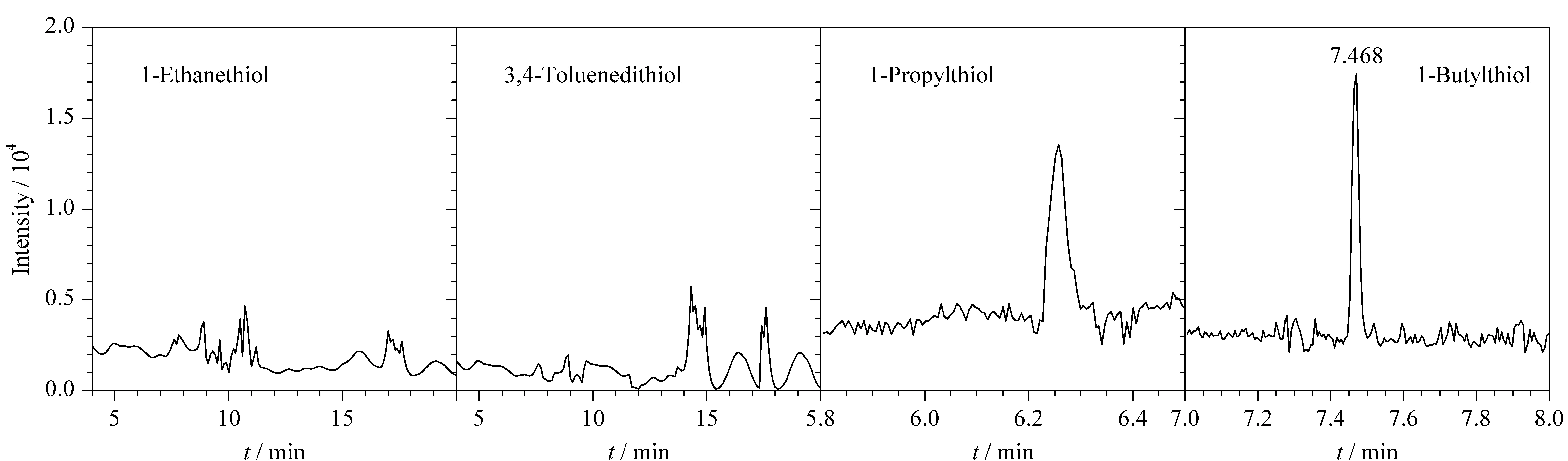
圖 2 采用不同衍生試劑時(shí)ClCN衍生產(chǎn)物的色譜圖Fig. 2 Chromatograms of ClCN derivative products with different derivatization reagents
2.3 衍生條件的優(yōu)化
分別配制不同有機(jī)溶劑的ClCN(1 mg/L)標(biāo)準(zhǔn)工作溶液,采用1-丁基硫醇作為衍生化試劑,設(shè)計(jì)四因素三水平的正交試驗(yàn),每組實(shí)驗(yàn)平行重復(fù)3次,結(jié)果見表1。根據(jù)顯著性分析結(jié)果可知,在所選因素水平范圍內(nèi),影響衍生結(jié)果的顯著性次序?yàn)?體系pH值>衍生溫度>衍生時(shí)間>溶劑體系,其中只有體系pH值對(duì)實(shí)驗(yàn)結(jié)果有顯著性影響。當(dāng)反應(yīng)體系的pH=9時(shí),ClCN衍生產(chǎn)物的色譜峰面積要明顯高于中性及酸性體系中衍生產(chǎn)物的峰面積。
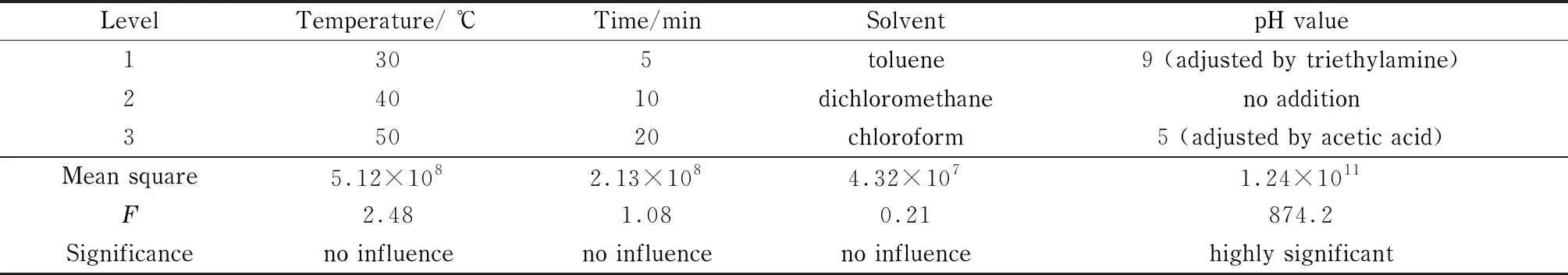
表 1 正交試驗(yàn)結(jié)果
根據(jù)衍生反應(yīng)方程式可知,在體系中加入縛酸劑能夠有效促進(jìn)衍生反應(yīng)向正反應(yīng)方向進(jìn)行。考慮到時(shí)間成本以及合適的溫度,最優(yōu)衍生條件為:反應(yīng)溫度40 ℃,衍生時(shí)間10 min,反應(yīng)體系pH=9,有機(jī)相和水相分別采用三乙胺及氫氧化鈉作為縛酸劑。2.4 頂空-固相微萃取條件優(yōu)化
考察不同頂空-固相微萃取溫度對(duì)實(shí)驗(yàn)結(jié)果的影響。在室溫(25 ℃)下對(duì)含500 μg/L ClCN/去離子水進(jìn)行巰基化衍生后,在不同的溫度(25、35、45、55、65、75、85 ℃)下進(jìn)行頂空-固相微萃取吸附。每個(gè)溫度下進(jìn)行3次重復(fù)試驗(yàn),比較不同頂空溫度下ClCN衍生產(chǎn)物的峰面積(見圖3)。
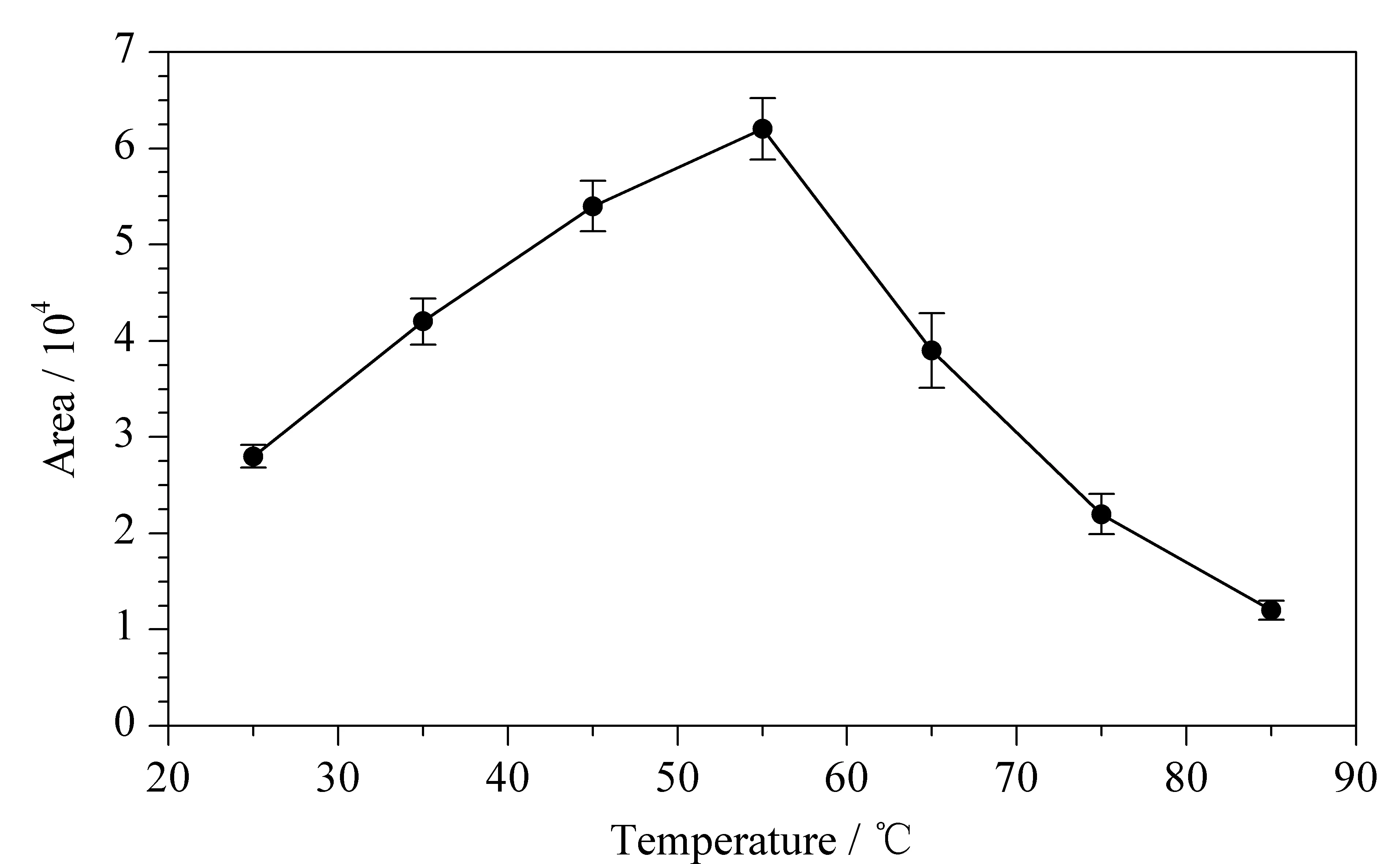
圖 3 不同頂空-固相微萃取溫度對(duì)ClCN衍生產(chǎn)物峰面積的影響(n=3)Fig. 3 Effect of different temperatures for HS-SPME on the peak area of derivative of ClCN (n=3)
結(jié)果表明,當(dāng)頂空溫度為25~55 ℃時(shí),目標(biāo)物隨著頂空溫度的上升,峰強(qiáng)度增加。當(dāng)頂空溫度超過55 ℃時(shí),目標(biāo)物峰面積減小,表明頂空溫度的進(jìn)一步升高對(duì)固相微萃取過程不利。可能原因是升溫有助于目標(biāo)物的揮發(fā),而溫度過高會(huì)導(dǎo)致固相微萃取纖維的解吸,不利于目標(biāo)物在萃取纖維上的富集。因此本實(shí)驗(yàn)選擇55 ℃作為頂空-固相微萃取溫度。
500 μL 10 mg/L ClCN/去離子水溶液采用固相微萃取進(jìn)行樣品制備及儀器分析時(shí),目標(biāo)化合物的保留時(shí)間與有機(jī)相時(shí)不同(見圖4),為6.844 min,原因在于萃取纖維的吸附/解吸速率與直接進(jìn)樣的氣化速度不同。
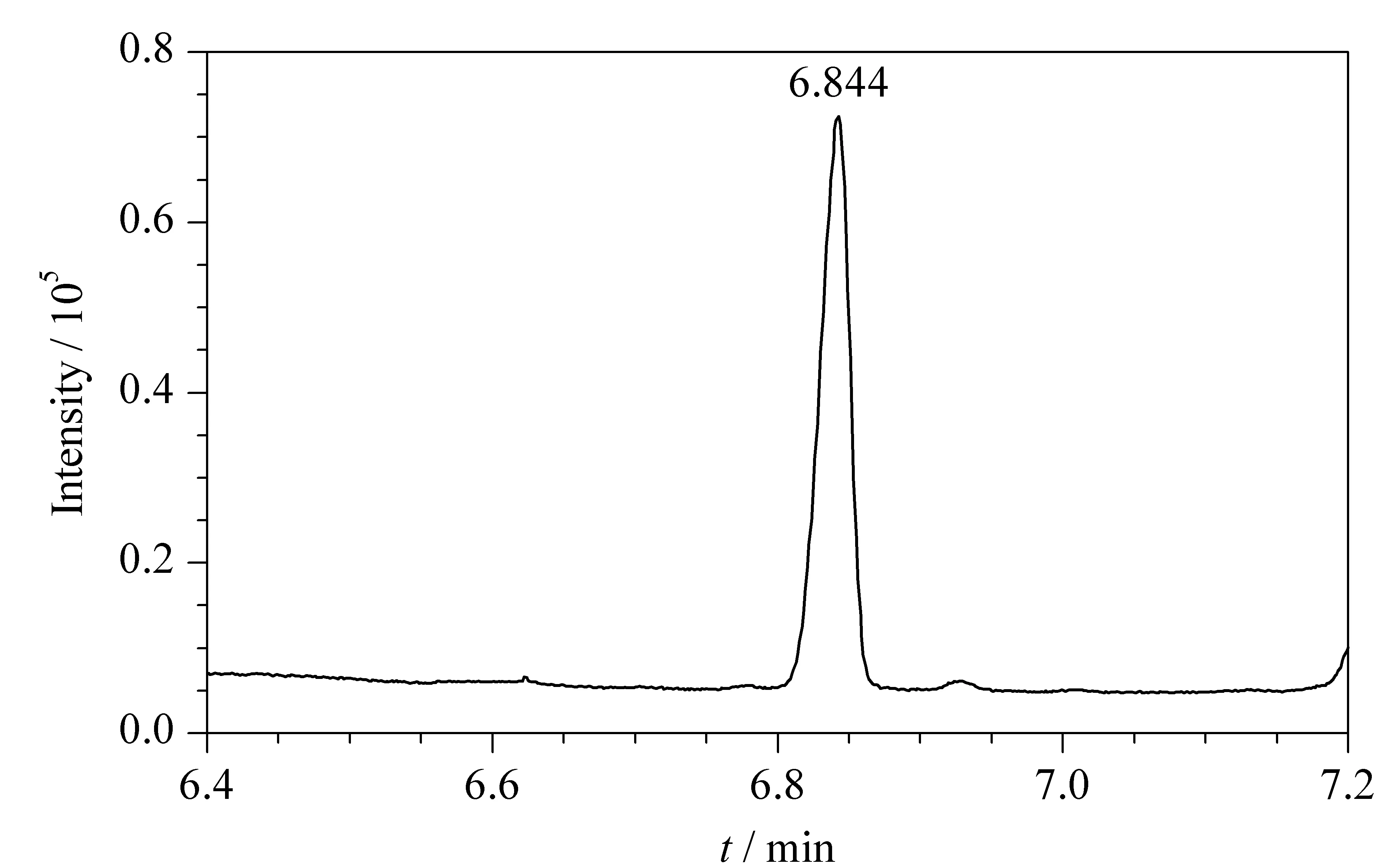
圖 4 水樣基質(zhì)中10 mg/L ClCN衍生產(chǎn)物的色譜圖Fig. 4 Chromatogram of derivative of ClCN at 10 mg/L in the water matrix
2.5 衍生產(chǎn)物的鑒定及質(zhì)譜裂解規(guī)律的分析
根據(jù)ClCN巰基化衍生產(chǎn)物的反應(yīng)式和GC-EI/MS質(zhì)譜圖,可知衍生產(chǎn)物為硫氰酸丁酯,主要質(zhì)譜碎片離子的m/z為29、41、57、115等。
進(jìn)一步對(duì)硫氰酸丁酯的碎片離子峰進(jìn)行歸屬并推斷其質(zhì)譜裂解規(guī)律:m/z為115的碎片離子峰代表衍生產(chǎn)物硫氰酸丁酯的分子離子峰,m/z為57和29的碎片離子分別代表硫氰酸丁酯經(jīng)電子轟擊電離后產(chǎn)生的丁烷基和乙烷基碎片離子。但是質(zhì)譜圖中m/z為41的碎片離子無法直接歸屬。
采用GC-MS/MS子離子掃描的方式進(jìn)一步對(duì)分子碎片進(jìn)行碰撞解析。首先選擇m/z為115的質(zhì)譜碎片作為母離子,碰撞能量設(shè)為10 eV。在子離子的質(zhì)譜圖中發(fā)現(xiàn)了m/z分別為115、57、41、和29的碎片離子。該結(jié)果證明,m/z為29、41、57的碎片離子由衍生產(chǎn)物硫氰酸丁酯的分子離子經(jīng)碰撞裂解后所產(chǎn)生。
進(jìn)一步選擇m/z為57的碎片離子作為母離子,在不同的碰撞能量(5、10及20 eV)下進(jìn)行裂解。如圖5所示,不同的碰撞能量下均可發(fā)現(xiàn)m/z為41的質(zhì)譜碎片。該結(jié)果證明,m/z為41的質(zhì)譜碎片可由m/z為57的質(zhì)譜碎片進(jìn)一步碰撞裂解得到,而并非來自硫氰根(-SCN)。這與烷烴質(zhì)譜的裂解規(guī)律相符,m/z為41的碎片離子來自于碳正離子的裂解,符合偶電子規(guī)律。同時(shí)根據(jù)質(zhì)譜豐度可知5 eV的碰撞能量最佳。
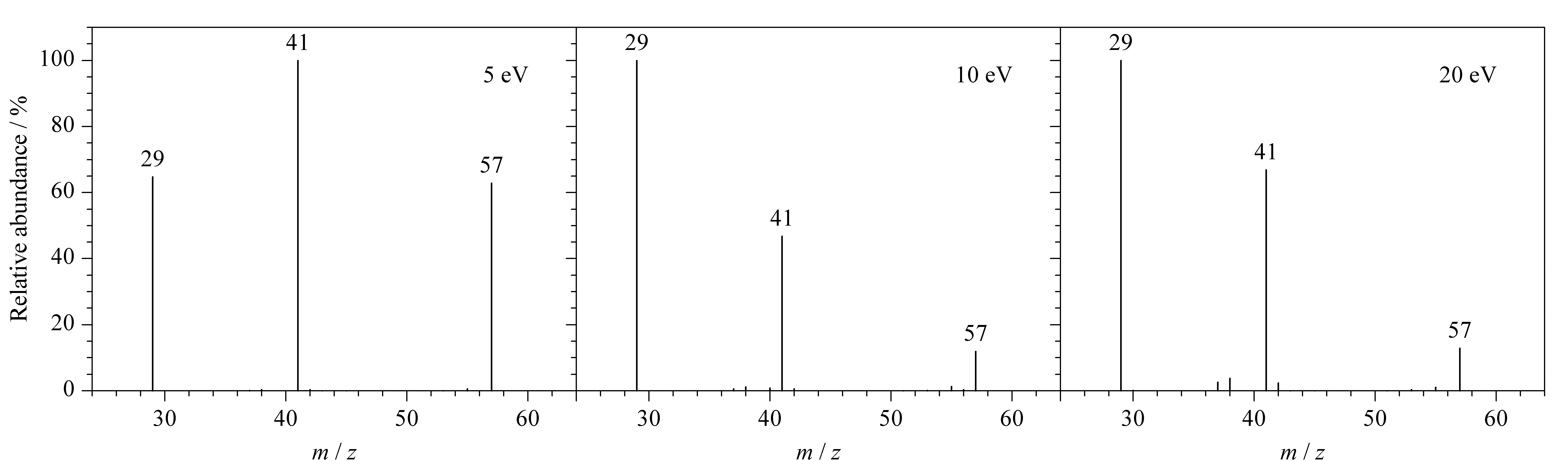
圖 5 不同碰撞能量下m/z 57的碎片離子作為母離子時(shí)的質(zhì)譜圖Fig. 5 Mass spectra with fragment ions of m/z 57 as the precursor ions under different collision energies
根據(jù)GC-MS/MS結(jié)果,對(duì)所有的碎片離子進(jìn)行歸屬并推斷硫氰酸丁酯在電子轟擊電離下的裂解途徑。如圖6所示,推斷m/z=41的離子碎片為烯丙基碳正離子結(jié)構(gòu)。
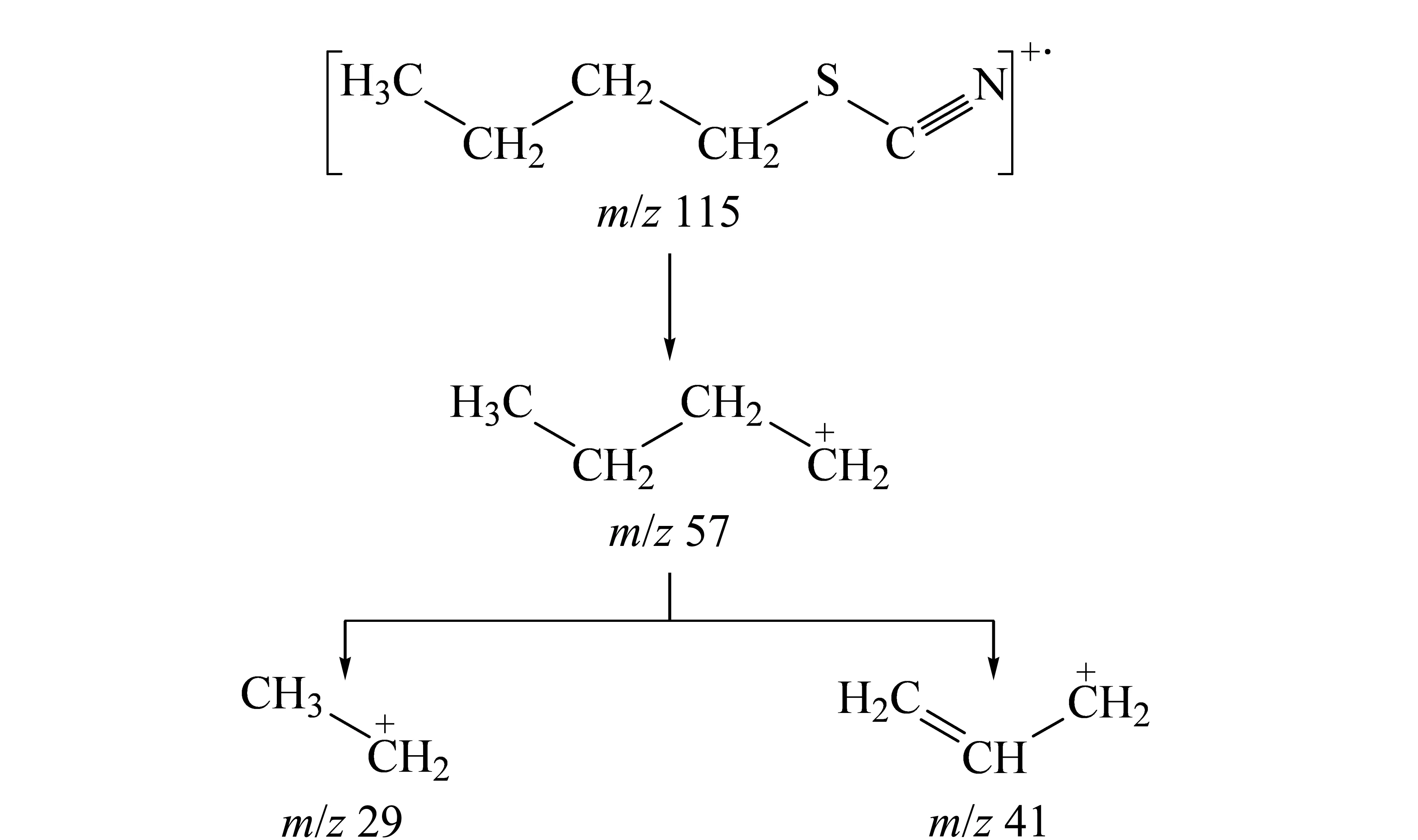
圖 6 硫氰酸丁酯的質(zhì)譜裂解規(guī)律Fig. 6 MS fragmentation route of butyl thiocyanate
通過對(duì)ClCN衍生產(chǎn)物質(zhì)譜圖中主要碎片離子的分析,能夠推斷化合物在電子轟擊條件下的裂解途徑,有助于理解并闡明主要質(zhì)譜碎片之間的相互關(guān)系,并為未來建立痕量ClCN的GC-MS/SRM(選擇反應(yīng)監(jiān)測(cè)掃描)方法提供技術(shù)儲(chǔ)備。
2.6 定量方法的選擇
采用內(nèi)標(biāo)法進(jìn)行定量測(cè)定能夠避免實(shí)驗(yàn)前處理操作中目標(biāo)成分丟失造成的定量不準(zhǔn)確,還能有效提高檢測(cè)靈敏度并降低樣品制備過程中的干擾。本研究在實(shí)驗(yàn)條件允許的情況下,采用內(nèi)標(biāo)法對(duì)有機(jī)相中ClCN進(jìn)行定量分析,以獲得更好的定量準(zhǔn)確度。
異丙基二硫醚具有較好的化學(xué)穩(wěn)定性,方便保存,且高純度標(biāo)準(zhǔn)品易于購買。同時(shí)異丙基二硫醚與衍生產(chǎn)物均含S元素,化學(xué)性質(zhì)較為類似。在本研究中的色譜及質(zhì)譜條件下,異丙基二硫醚的保留時(shí)間(8.050 min)與目標(biāo)物接近(7.468 min),且色譜峰能夠完全分離。因此,選擇異丙基二硫醚作為內(nèi)標(biāo)物進(jìn)行有機(jī)相中ClCN的定量分析。
水相中采用了頂空-固相微萃取的方式對(duì)ClCN衍生產(chǎn)物進(jìn)行富集,如果在樣品中加入內(nèi)標(biāo)物,在頂空加熱過程中內(nèi)標(biāo)物會(huì)與ClCN衍生產(chǎn)物在固相微萃取纖維上發(fā)生競(jìng)爭(zhēng)性吸附,從而影響檢測(cè)靈敏度以及實(shí)驗(yàn)結(jié)果的精密度。因此水相中的ClCN采用外標(biāo)法定量。
2.7 方法學(xué)考察
2.7.1線性范圍,檢出限和定量限
分別配制10~2 000 μg/L ClCN/甲苯標(biāo)準(zhǔn)工作溶液及10~1 200 μg/L ClCN/去離子水標(biāo)準(zhǔn)工作溶液,依次根據(jù)1.2節(jié)和1.3節(jié)進(jìn)行制備和測(cè)定。
在有機(jī)相中選擇異丙基二硫醚為內(nèi)標(biāo)物,以ClCN的質(zhì)量濃度為橫坐標(biāo)(x, μg/L)、ClCN衍生產(chǎn)物與內(nèi)標(biāo)物峰面積之比為縱坐標(biāo)(y1)進(jìn)行線性擬合。對(duì)于水相中ClCN的測(cè)定,以ClCN的質(zhì)量濃度為橫坐標(biāo)(x, μg/L), ClCN衍生產(chǎn)物峰面積為縱坐標(biāo)(y2)進(jìn)行線性擬合。
以響應(yīng)顯著高于溶劑空白且信噪比為3對(duì)應(yīng)的濃度作為方法的檢出限(LOD),以線性范圍最低點(diǎn)的濃度作為方法的定量限(LOQ)。有機(jī)相及水相中ClCN的線性方程、檢出限和定量限結(jié)果見表2。

表 2 不同基質(zhì)中ClCN的線性范圍、線性方程、相關(guān)系數(shù)(R2)、檢出限及定量限
2.7.2加標(biāo)回收率與精密度
以甲苯及不同來源的水樣(去離子水、河水、自來水、飲用水)作為樣品基質(zhì),分別對(duì)制3個(gè)水平的ClCN基質(zhì)溶液進(jìn)行加標(biāo)回收試驗(yàn),每個(gè)水平平行測(cè)定6次(見表3)。結(jié)果表明,在甲苯中ClCN的加標(biāo)回收率為87.3%~98.8%, RSD為2.1%~4.7%;在不同的水樣基質(zhì)樣本中ClCN的加標(biāo)回收率為97.6%~102.2%, RSD為2.8%~4.2%。表明本方法的穩(wěn)定性和重復(fù)性均較好。
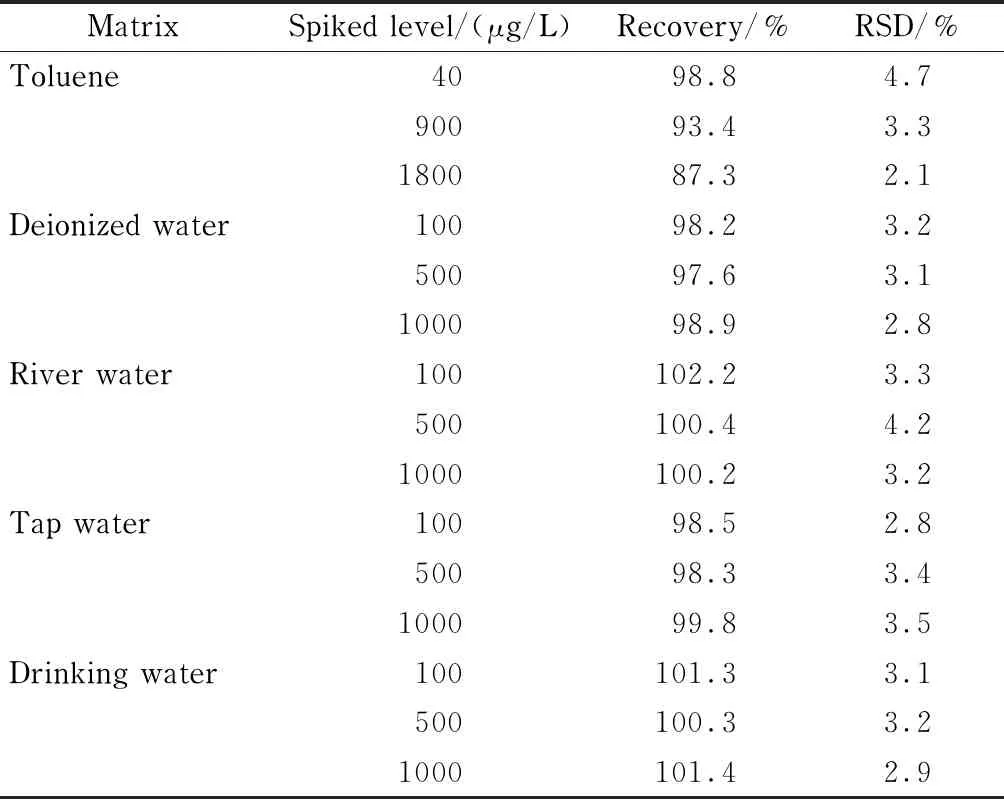
表 3 不同基質(zhì)中ClCN的加標(biāo)回收率和相對(duì)標(biāo)準(zhǔn)偏差(n=6)
2.8 實(shí)際樣品測(cè)定
本實(shí)驗(yàn)室為禁止化學(xué)武器組織(OPCW)指定實(shí)驗(yàn)室,參與OPCW第33次水平考試的配樣過程。取第33次水平考試的空白有機(jī)樣品(樣品基質(zhì)為正己烷,樣品編號(hào)333),加入ClCN標(biāo)準(zhǔn)工作溶液,使其最終質(zhì)量濃度為1 000 μg/L(選擇該濃度的原因是OPCW水平考試中規(guī)定目標(biāo)化合物最低添加含量為1 000 μg/L)。取該樣品1.0 mL,根據(jù)已建立的方法測(cè)定樣品中CNCl的含量,結(jié)果為982.3 μg/L,RSD為1.13%(n=3)。表明采用本實(shí)驗(yàn)建立的方法能夠有效測(cè)定模擬加標(biāo)實(shí)際樣品中CNCI的含量。
3 結(jié)論
本研究建立了巰基化衍生-氣相色譜-質(zhì)譜測(cè)定氯化氰的方法,并對(duì)衍生產(chǎn)物的質(zhì)譜裂解規(guī)律進(jìn)行了探究。針對(duì)不同溶劑的樣本,分別通過直接衍生及頂空-固相微萃取的方式進(jìn)行了樣品制備過程。本方法靈敏度高,選擇性好,能夠?qū)崿F(xiàn)不同基質(zhì)樣本中氯化氰的定性分析及定量測(cè)定。
