高效液相色譜-氣相色譜在線聯用同時測定土壤中飽和烴和芳香烴
2021-07-01 03:54:42劉玲玲李冰寧武彥文
色譜 2021年8期
關鍵詞:分析
劉玲玲, 李冰寧, 武彥文
(北京市理化分析測試中心, 北京市食品安全測試工程技術研究中心, 北京 100094)
石油作為能源和化學工業的原材料在全球的應用十分普遍,然而,其開采、運輸、儲存等過程均可能對環境造成污染風險,此外,一些人為因素導致的環境污染也不容忽視,如油罐泄露、油輪事故、廢水排放等[1]。烴類污染物不易降解,會持續影響生態系統和人類健康。因此,土壤中的石油污染一直是環境監測的重點[2,3]。
石油的化學組成非常復雜,包含不同結構類型的碳氫化合物,即烴類化合物。石油烴的結構類型包括直鏈或支鏈脂肪族、脂環族和高度烷基化的芳香族,其中,直鏈或支鏈脂肪族、脂環族統稱為飽和烴(saturated hydrocarbons, SH),含有烷基化芳香族則統稱為芳香烴(aromatic hydrocarbons, AH)[4,5]。由于石油烴的碳數從十幾到幾十,因此其包含的化合物數量巨大,無法將其分離成單個物質分析,只能進行總量測定。紅外光譜法、紫外和熒光分光光度法等分子光譜分析方法均曾經用于石油烴的總量測定[6-9],但均存在局限性。紫外和熒光方法只能測定芳香烴,紅外光譜法雖然可以同時測定飽和烴和芳香烴,但需要使用劇毒的四氯化碳或四氯乙烯。更為關鍵的是,由于石油烴的污染來源復雜且多樣,不同樣品中石油烴的化學組成迥異,無法找到合適的標準品與之匹配。分子光譜法缺乏與目標物匹配的標準品,其測定結果無法真實反映實際情況。氫火焰離子化檢測器(FID)對烴類物質的響應幾乎完全一致,因而其定量結果與標準品無關,只需任一烴類化合物作為內標即可對不同來源的石油烴污染物進行準確定量[10]。因此,近年來氣相色譜法(GC)成為環境中石油烴分析的新標準[11-16]。
然而,GC-FID缺乏選擇性,無法區分飽和烴與芳香烴,石油烴中不同組分的毒性差別很大,如飽和烴有蓄積作用,導致微型肉芽腫的形成,而芳香烴可能致畸和致癌風險[17]。為了分別測定飽和烴和芳香烴,美國馬薩諸塞州的標準方法是將樣品通過二氯甲烷萃取,經硅膠柱分離得到飽和烴和芳香烴[5]。不過,該方法消耗溶劑多、步驟繁瑣、操作難度較大。目前,食品領域采用在線聯用高效液相色譜-氣相色譜(on-line high performance liquid chromatography-gas chromatography, HPLC-GC)技術測定其中的礦物油污染物,該技術有效整合了烴類分析中的凈化、分離與檢測步驟,其中HPLC的硅膠柱可以吸附油脂等極性物質,同時分離飽和烴和芳香烴,然后通過閥切換、預柱與溶劑排空閥組成的HPLC-GC接口,將分離出來的飽和烴和芳香烴全部送入GC分析,真正實現了全樣品分析,從而極大地提升了分析靈敏度;此外,這套系統通常配置兩套完全相同的通道(每個通道均由一套預柱、三通、溶劑放空閥、分析柱和FID組成),一次進樣即可同時實現飽和烴和芳香烴的測定,成倍地提高了分析效率。HPLC-GC的應用有效減少了樣品量和試劑消耗,簡化了實驗步驟,避免了污染引入,從而提高了方法的準確性和結果的重現性[10,18]。我們前期將該技術成功運用于奶粉、大米、巧克力等食品中礦物油的測定[19-21],結果發現,不同樣品的基質干擾不同,其涉及的提取與凈化方法各有差異。以提取方法為例,大部分干物質可以直接用正己烷浸泡提取,濕物質則需要事先去除水的干擾,而一些噴霧干燥的樣品則需要水解處理才能提取完全[22],等等。本研究將HPLC-GC用于土壤中飽和烴與芳香烴的測定,需要根據土壤基質的干擾情況對樣品前處理方法進行優化。首先本文對提取方法進行了考察和優化,包括提取溶劑、時間、溫度和次數等提取條件;其次,由于HPLC-GC中HPLC硅膠柱的吸附容量有限,本文增加離線的固相萃取(SPE)凈化步驟,考察優化了去除提取液中油脂等極性干擾物的凈化條件;最后,采用HPLC-GC技術建立了同時測定土壤中飽和烴和芳香烴的方法。此外,本文還通過譜圖分析探索了石油烴的污染來源,這些均為了解環境中石油烴的污染情況與制定治理策略提供數據支撐。
1 實驗部分
1.1 儀器、試劑與材料
HPLC-GC聯用儀器:包括配備二元泵和UV檢測器的LC 20A液相色譜儀,帶有FID的GC 2010 plus氣相色譜儀(日本Shimadzu公司), HPLC-GC接口(德國Axel Semrau公司)和PAL自動進樣器(瑞士CTC公司)。
正己烷、二氯甲烷、無水乙醇、甲苯均為色譜純(美國Fisher Scientific公司);無水硫酸鈉為分析純(國藥集團化學試劑有限公司);硅膠(0.063~0.200 mm),使用前400 ℃下活化16 h(德國Merck公司)。
SQC-116的土壤石油烴標準物(標準值2 541 mg/kg,可信區間938~3 750 mg/kg)購自美國NSI Lab solutions公司;9種飽和烴/芳香烴混合標準溶液(15~60 mg/L,溶劑是正己烷和/或甲苯,分別作為內標使用):正十三烷(n-C13)的質量濃度為15 mg/L,正十一烷(n-C11)、環己基環己烷(Cycy)、戊基苯(5B)、1-甲基萘(1-MN)、2-甲基萘(2-MN)、1,3,5-三叔丁基苯(TBB)的質量濃度均為30 mg/L, 5α-膽甾烷(Cho)和苝(Per)的質量濃度均為60 mg/L,以上9種內標物質以及C7~C40正構烷烴混合標準品(1 000 mg/L)均購自美國Sigma-Aldrich公司。其中n-C11和5B分別用于飽和烴和芳香烴部分的揮發損失考察;Cycy和2-MN分別為飽和烴和芳香烴的定量內標;n-C13和1-MN用于考察內標物Cycy和2-MN的響應信號的準確性;此外,Cho、TBB和Per用于監控HPLC分離情況,它們分別為飽和烴流分的末端、芳香烴的開端與末端標記物。
1.2 試樣制備
1.2.1試樣提取
稱取5.0 g土壤樣品,加入20 mL正己烷-乙醇(1∶1, v/v)混合試劑和30 μL飽和烴/芳香烴混合標準溶液,振蕩1 h,然后加入20 mL去離子水,水洗去乙醇,取正己烷相。
1.2.2試樣凈化
將2 g活化硅膠裝入玻璃層析柱,硅膠頂部覆蓋1 g無水硫酸鈉,以10 mL正己烷-二氯甲烷混合溶劑(8∶2, v/v)平衡柱床,然后加入上述提取液,待液面近干,以10 mL左右的正己烷-二氯甲烷混合溶劑(8∶2, v/v)淋洗,收集洗脫液并濃縮至約1 mL,注入HPLC-GC分析。
1.3 HPLC-GC分析
1.3.1HPLC條件
Allure Si色譜柱(250 mm×2.1 mm, 5 μm, 6 nm,美國Restek公司);流動相A為正己烷,B為二氯甲烷;梯度洗脫:0~0.1 min, 100%A(流速為0.3 mL/min); 0.1~6.2 min, 70%A(流速為0.3 mL/min); 6.2~15.2 min, 100%B(反沖,流速為0.5 mL/min); 15.2~25.2 min, 100%A(流速為0.5 mL/min); 25.2~30 min, 100%A(流速為0.3 mL/min)。HPLC運行過程的流動相變換與飽和烴/芳香烴流出通過紫外檢測器(230 nm)監測,進樣量為50 μL。
1.3.2HPLC-GC接口
基于HPLC-GC聯用技術[21],經HPLC分離后分別得到450 μL的飽和烴(2.0~3.5 min)和芳香烴(4.5~6.0 min)。兩段流分通過閥切換,以氫氣為載氣被導入GC分析。GC儀器配備了由預柱(Restek MXT無涂層毛細管預柱,10 m×0.53 mm)和分析柱(Restek MXT毛細管柱,15 m×0.25 mm×0.25 μm)組成的兩個平行通道。飽和烴和芳香烴各自進入一個通道,預柱與分析柱之間通過三通與溶劑排空閥連接。溶劑排空閥在LC流分閥切換前0.5 min開啟,轉移結束后0.3 min關閉。轉移到GC系統的飽和烴和芳香烴中大部分溶劑通過溶劑排空閥去除,剩余少量溶劑與濃縮的溶質聚集在分析柱入口,進行后續GC分離和測定。
1.3.3GC條件
程序升溫的初始溫度60 ℃(保持6 min),以15 ℃/min升溫至120 ℃,再以25 ℃/min升溫至370 ℃(保持6 min)。FID溫度為380 ℃;輔助氣、燃燒氣和助燃氣分別為氮氣、氫氣和空氣,流速分別為30、40和400 mL/min。
1.4 數據分析
飽和烴和芳香烴在GC-FID譜圖中均呈一定沸程范圍的駝峰,定量計算時,飽和烴/芳香烴的含量通過計算譜圖基線與駝峰之間的面積得到,基線取決于樣品空白的GC譜圖;駝峰上方的內標物尖峰定量計算時扣除[10]。采用內標法對飽和烴/芳香烴進行定量,飽和烴的定量內標物為環己基環己烷(Cycy),芳香烴的定量內標物為2-MN。
1.5 空白實驗
除不加土壤樣品外,按照1.2節和1.3節進行操作,得到樣品空白實驗的HPLC-GC譜圖,該譜圖不含干擾石油烴測定的駝峰,基線近似平直,基線偏移的高度與石油烴信號的高度之比不超過四分之一。
2 結果與討論
2.1 提取
2.1.1提取溶劑的選擇
根據相似相溶原理,正己烷、正庚烷、二氯甲烷等弱極性溶劑可用于提取石油烴。由于土壤中含有不同程度的水分,土壤顆粒孔隙中的水會阻止石油烴擴散至提取溶劑,因而需要對濕樣品進行事先脫水。然而,通常采用的蒸發脫水容易導致揮發性烴類物質的損失。因此,通過加入與水互溶的極性溶劑,利用其滲透固體顆粒表面的水層來促進濕樣品中石油烴的溶出。國際標準ISO 16703采用丙酮-正庚烷(2∶1, v/v)或非極性溶劑(如石油醚、環己烷、正己烷等)提取土壤中的石油烴[4];美國標準EPA 3540C規定土壤/沉積物的提取溶劑采用正己烷-丙酮(1∶1, v/v)或二氯甲烷-丙酮(1∶1, v/v)[23];我國環境行業標準HJ 1021-2019《土壤沉積物石油烴(C10~C40)的測定 氣相色譜法》選用正己烷-丙酮(1∶1, v/v)或正己烷作為提取溶劑[16]。
由于正庚烷的沸點較高,為98.5 ℃,去除溶劑時需要的時間較長;丙酮屬于易制毒品,購買受到限制;二氯甲烷則被列入《優先控制化學品名錄》,應最大限度減少生產和使用。因此,選取正己烷替代正庚烷和二氯甲烷,以乙醇替代丙酮作為石油烴提取溶劑。本文分別考察和對比了正己烷和正己烷-乙醇(1∶1, v/v)的提取效果。結果表明:正己烷-乙醇(1∶1, v/v)的提取效率(13.9 mg/kg)明顯優于正己烷的(5.8 mg/kg)(色譜圖見圖1);同時乙醇的毒性很低,故本研究選取正己烷-乙醇(1∶1, v/v)作為土壤中石油烴的提取溶劑。
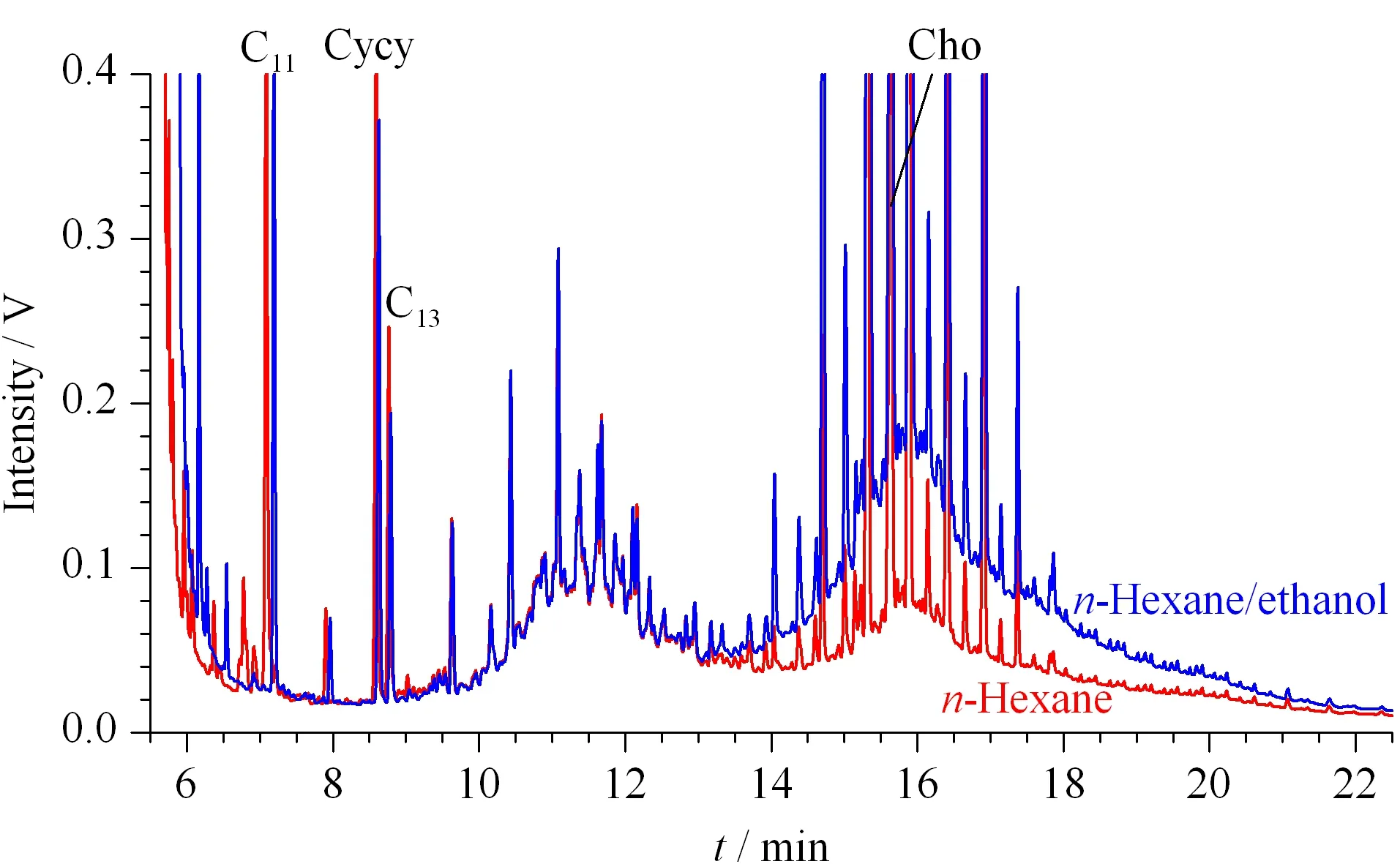
圖 1 不同溶劑提取同一土壤樣品中飽和烴的HPLC-GC譜圖Fig. 1 HPLC-GC chromatograms of saturated hydrocarbons (SH) in the same soil sample extracted using different solvents Cycy: cyclohexylcyclohexane; Cho: 5α-cholestane.
2.1.2提取級數的優化
為了考察提取是否完全,將土壤樣品按照2.1.1節的方法再重復提取一次,然后合并兩次提取液。結果發現:提取兩次比提取一次的時間和溶劑消耗量均增加一倍,但飽和烴的含量僅增加了4.3%(見表1),說明經過一次提取即可將絕大部分的石油烴提取出來。因此,確定提取級數為一次。
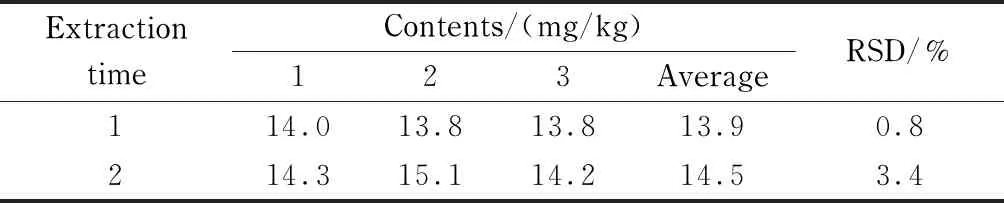
表 1 提取級數對同一土壤樣品中飽和烴(C10~C40)測定的影響
2.2 凈化
2.2.1凈化填料的選擇
提取石油烴時,土壤中的腐殖酸、脂類、色素等極性干擾物也被一同提取出來[24],這些干擾物影響后續的分析,因此,需要對提取液進行凈化。硅膠和硅酸鎂是常用的凈化填料[25],都能夠吸附動、植物油等極性干擾物質。本研究采用食品中礦物油分析常用的活化硅膠作為凈化填料[26-30],并且考察和優化了硅膠柱的凈化條件。
2.2.2洗脫溶劑的選擇
當石油烴提取液進入硅膠柱后,首先流出的是極性最低的飽和烴,隨后是極性稍高的芳香烴,最后是甘油三酯和環氧烯烴等極性更高一些的物質。具體細分到飽和烴部分,由于體積排阻效應,首先流出的是大相對分子質量的鏈烷烴、其次是低相對分子質量的鏈烷烴和環烷烴;對于芳香烴部分,則首先流出的是高度烷基取代的單環芳烴,其次是高度烷基取代的多環芳烴、最后是低烷基取代或無烷基取代的芳香烴。為了嚴格控制石油烴的凈化以及飽和烴和芳香烴的分離情況,通常以多個化合物標記組分的流出情況,即Cho標記飽和烴的末端,TBB和Per分別標記芳香烴的開端與末端。此外,標準溶液中通常還添加低沸點的n-C11和5B以分別考察飽和烴和芳香烴部分在整個前處理過程是否存在揮發損失;飽和烴部分的定量內標是Cycy(通常n-C13的濃度為Cycy的一半,用于考察Cycy的回收率),芳香烴部分的定量內標是2-MN或1-MN(兩種化合物的濃度相同)[18,31]。
由于HPLC硅膠色譜柱(250 mm×2.1 mm)只能吸附20 mg油脂[18],為了確保HPLC柱的分離性能,提取液在注入HPLC分離飽和烴和芳香烴之前需要預先采用SPE柱凈化。將1 mL飽和烴/芳香烴混合標準溶液轉移至SPE柱,待上樣液近干時,分別用15 mL正己烷和正己烷-二氯甲烷(8∶2, v/v)洗脫,每1 mL洗脫液收集1管,注入GC分析,考察洗脫溶劑能否將飽和烴和芳香烴完全洗脫。研究表明,僅用正己烷洗脫,直至最后1 mL芳香烴末端的標記物Per仍未流出;而采用正己烷-二氯甲烷(8∶2, v/v)洗脫,Per在第5管流出,11管結束(見圖2), 99%以上Per集中在5管到10管之間。為了充分回收芳香烴,本研究采用10 mL正己烷-二氯甲烷(8∶2, v/v)混合溶劑作為洗脫液。
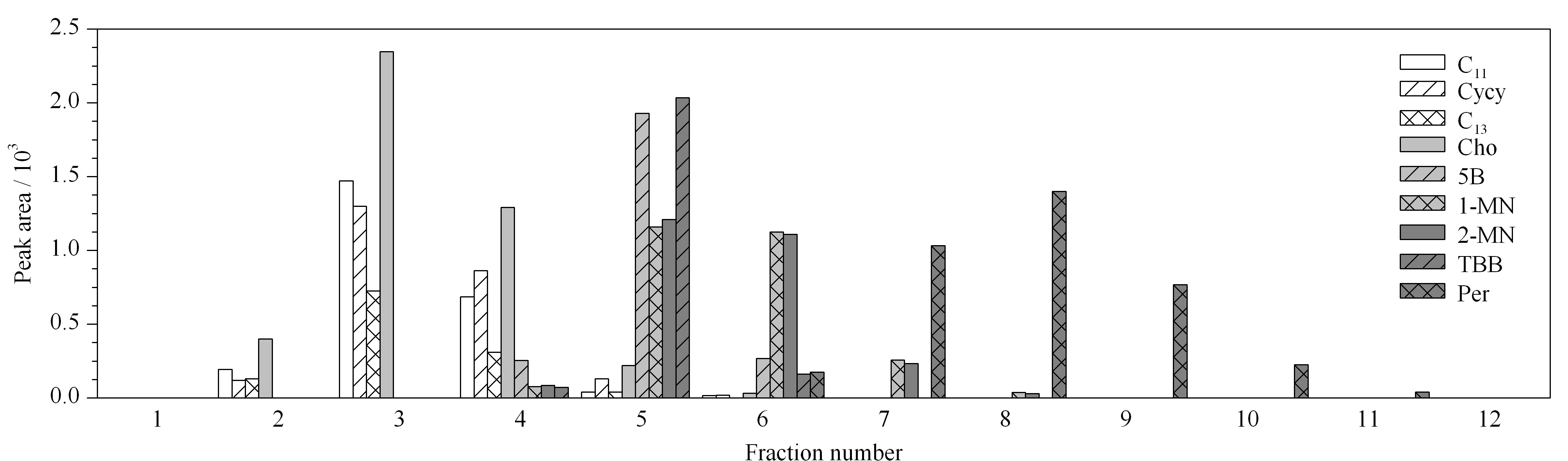
圖 2 飽和烴和芳香烴(AH)混合標準溶液通過硅膠柱凈化后以正己烷-二氯甲烷(8∶2, v/v)混合溶劑洗脫的結果Fig. 2 Elution order of mixed standard solution containing SH and aromatic hydrocarbons (AH) through the SPE column packed with silica gel and a solvent mixture of n-hexane and dichloromethane (8∶2, v/v)5B: pentylbenzene; MN: methylnaphthalene; TBB: tri-tert-butylbenzene; Per: perylene.
2.2.3HPLC-GC分析
將飽和烴/芳香烴混合標準溶液按照上述步驟凈化,所得洗脫液濃縮至約1 mL,注入HPLC-GC以1.3節條件分析。結果表明,n-C11、n-C13、Cycy和Cho只出現在飽和烴通道中,5B、1-MN、2-MN、TBB和Per只出現在芳香烴通道中;且n-C11和5B的回收率不低于90%, Cycy和n-C13的峰面積比例為2∶1, 1-MN和2-MN的峰面積比例為1∶1(見圖3)。說明SPE柱凈化性能和HPLC柱分離性能滿足同時測定土壤中飽和烴和芳香烴的需求。
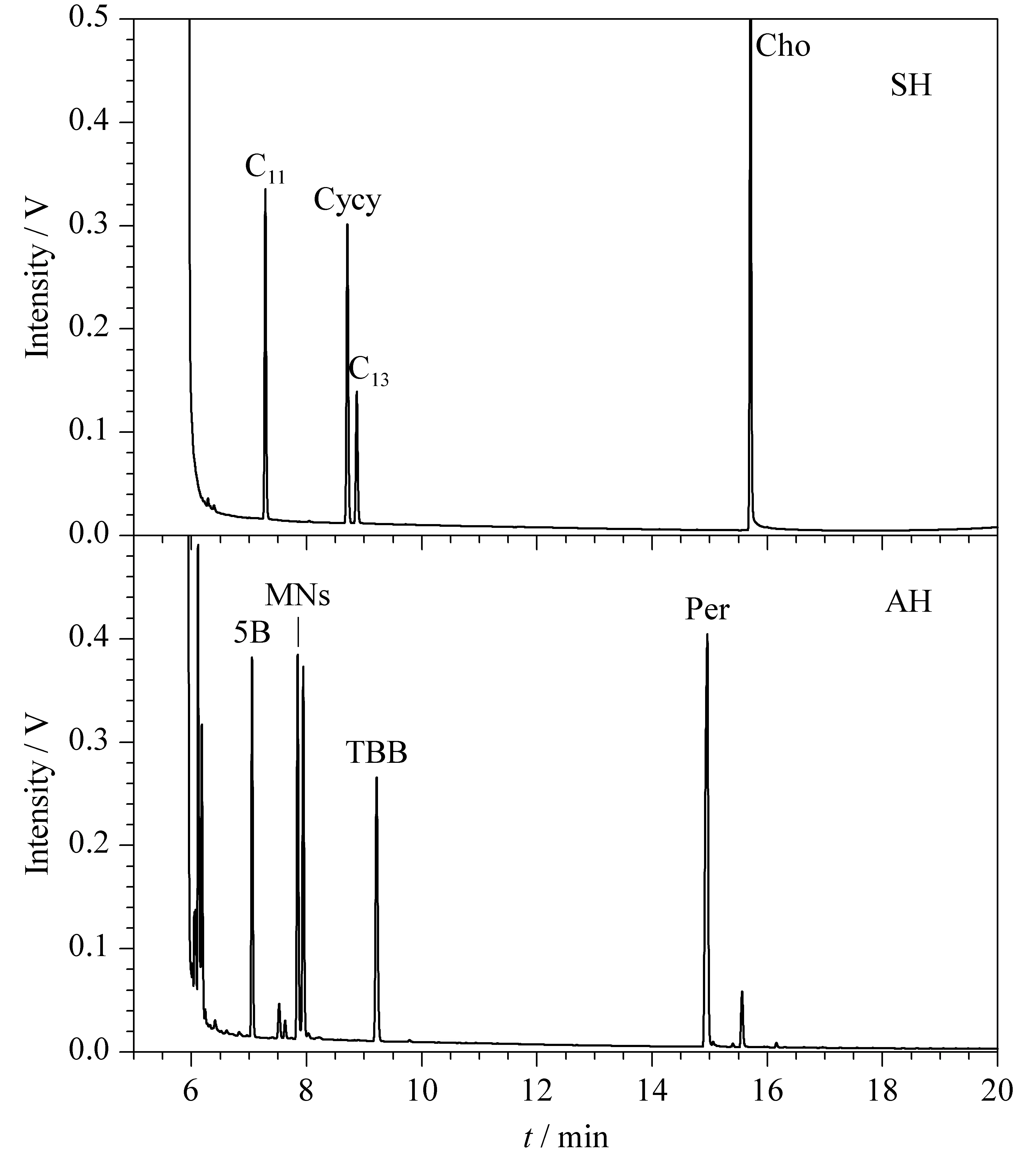
圖 3 SH/AH混合標準溶液經硅膠柱凈化后的HPLC-GC譜圖Fig. 3 HPLC-GC chromatograms of mixed standard solution of SH/AH purified by the SPE column packed with silica gel
2.3 方法學考察
2.3.1儀器性能考察
以1.3節條件分析n-C7~n-C40正構烷烴混合溶液(見圖4),n-C10、n-C11…、n-C39、n-C40相對于n-C20的響應因子在0.92~1.05之間,說明分析過程中低沸點目標物沒有揮發損失,對高沸點目標物沒有歧視,滿足分析要求(n-C10、n-C11、…、n-C39、n-C40相對于n-C20響應因子應高于0.80)[4]。
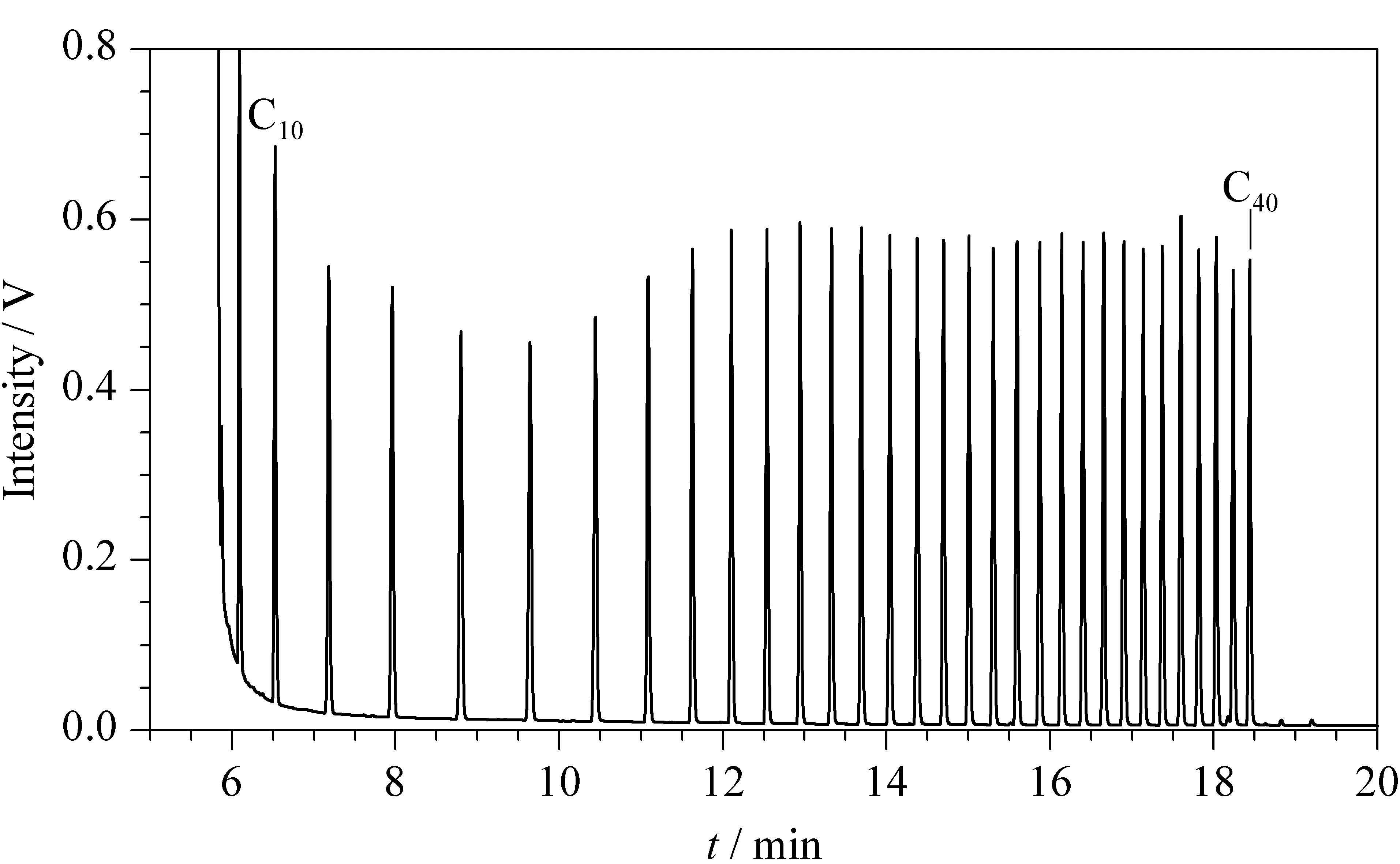
圖 4 n-C10~n-C40正構烷烴混合溶液的HPLC-GC譜圖Fig. 4 HPLC-GC chromatogram of n-alkane (C10-C40) standards
2.3.2線性范圍考察
分別配制2.5、4、8、20、50、150、250、500 mg/L潤滑油(飽和烴與芳香烴的質量比為85.5∶14.5)的正己烷溶液,取50 μL注入HPLC-GC測定。以潤滑油系列溶液濃度為橫坐標,分別以飽和烴和芳香烴駝峰的峰面積為縱坐標,繪制標準曲線。結果表明:飽和烴在2.1~427.5 mg/L、芳香烴在2.9~72.5 mg/L范圍內線性關系良好,線性方程分別為y=1 045.9x-1 532.3 (R2=0.999 7)和y=146.4x-772.3 (R2=0.999 3)。
2.3.3定量限
由于石油烴的GC-FID譜圖呈駝峰,根據儀器噪聲計算檢出限的方法GB5009.1-2003不適用于本方法。本方法定量限采用文獻方法計算,即進入FID的石油烴總量至少達到100 ng才能準確測定[18]。本方法稱取5 g土壤,經提取、SPE柱凈化、濃縮等操作后得到體積為1 mL的供試液,最后取50 μL注入HPLC-GC定量分析,即相當于有0.25 g樣品中的石油烴進入FID檢測,因而對應的LOQ值為0.4 mg/kg。
2.3.4準確度和精密度驗證
稱取0.5 g SQC-116土壤石油烴標準物質7份,用上述方法測定,考察方法的準確度和精密度。測定平均值為2 809 mg/kg,與標準值相對誤差(RE)為10.6%,在證書提供的可信區間內;相對標準偏差(RSD)為1.4%,表明此方法分析測定土壤中的石油烴具有良好的精密度和準確度。
2.4 實際樣品分析
本實驗采用上述方法,檢測了北京地區的5個土壤樣品中飽和烴和芳香烴含量,結果見表2。結果表明,5個土壤樣品中均檢出飽和烴(C10~C40),其含量范圍為3.3~32.1 mg/kg;其中4個樣品中檢出芳香烴(C10~C40),其含量范圍為0.8~4.3 mg/kg,芳香烴的比例范圍為6.3%~13.2%。
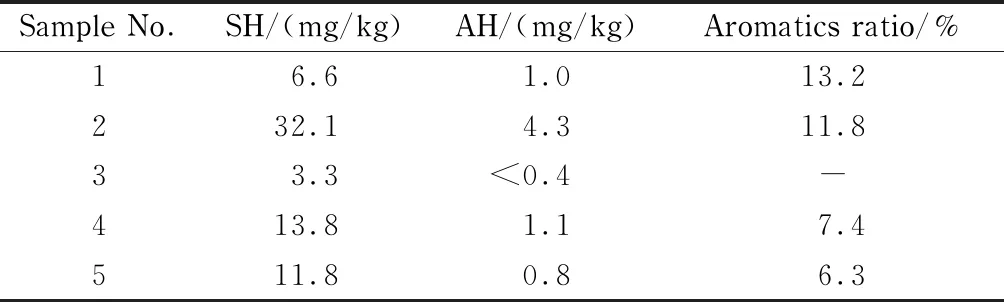
表 2 土壤樣品中飽和烴和芳香烴(C10~C40)的含量
我們分析了兩個土壤樣品的譜圖(見圖5)。結果顯示:不同樣品的飽和烴與芳香烴呈現出不同的碳數范圍分布,其中No.1樣品中的飽和烴由兩個駝峰組成,低碳數范圍以n-C18為中心,含量為3.0 mg/kg,高碳數范圍以n-C29為中心,含量為3.6 mg/kg(圖5a-SH);此外,No.1樣品的芳香烴譜圖也顯示出與低碳數飽和烴(以n-C18為中心)對應的駝峰,含量為0.6 mg/kg(圖5a-AH)。同樣,No.5土壤樣品也顯示出同樣碳數范圍的飽和烴和芳香烴駝峰(圖5b),只是含量不同。通常認為低碳數范圍的烴類物質主要來自柴油,而高碳數的烴類物質駝峰是潤滑油的典型特征[32]。圖5的結果表明:土壤中的烴類物質來源復雜,包含不同的碳數范圍,其中低碳數范圍含有芳香烴污染物。
3 結論
本研究建立了高效液相色譜-氣相色譜聯用法同時測定土壤中飽和烴和芳香烴的方法,針對土壤樣品特性,優化了提取和凈化兩個試樣制備環節。該方法整合了石油烴分析的凈化、分離與檢測步驟,靈敏度高、溶劑消耗少、準確度及精密度良好,適合于土壤中可萃取性石油烴的測定。同時,通過HPLC柱有效分離飽和烴和芳香烴,實現了毒性相對較高的芳香烴的測定,能更好地滿足石油烴污染物的監測與評估需要。此外,通過譜圖分析還可以探索石油烴的污染來源,有助于進一步提出相應的預防處理措施。
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
財經界(學術版)(2015年20期)2015-12-23 09:20:13
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
