化學交換法分離鋰同位素的理論及新技術研究進展
2021-06-30 01:29:08崔莉樊宇亨李莎莎白瑞兵郭彥霞程芳琴
化工學報 2021年6期
崔莉,樊宇亨,李莎莎,白瑞兵,郭彥霞,程芳琴
(1山西大學資源與環境工程研究所,國家環境保護煤炭廢棄物資源化高效利用重點實驗室,山西太原030006;2中國科學院過程工程研究所,中國科學院大學中丹學院,北京100049)
引 言
隨著人口增長和經濟的高速發展,能源短缺和生態環境問題日益凸顯,核能等新能源開發日趨重要。鋰(Li)是重要的能源戰略金屬,在自然界有6Li、7Li兩個穩定同位素,它們具有完全不同的核反應性[1]。其中6Li受中子轟擊可產生氚(6Li+n→T+4He),是核聚變反應必不可少的燃料,而氚是地球上最稀有的元素之一,其在自然界的豐度僅為0.004%。核聚變是一種更為清潔的核能源,是未來核能發展的趨勢,該過程要求6Li的豐度>30%。7Li俘獲中子的截面小,高純度的7Li(>99.995%)可作為核電站壓水堆的pH調節劑以及釷基熔鹽堆的冷卻劑。2012年,美國市場核電站壓水堆每年對7Li的需求量為400 kg,裝機容量為109W的釷基熔鹽反應器需要21~56 t的高純度7Li[1]。核能作為清潔新能源,我國早在2011年就啟動了戰略性先導科技專項“未來先進核裂變能”,如果可控核聚變得以實現,未來6Li、7Li需求量巨大[2]。6Li、7Li的天然豐度僅為7.42%和92.55%,因此,鋰同位素的分離和富集是核能開發必須解決的關鍵技術。
6Li、7Li具有完全相同的核外電子排布,化學性質非常相似,分離難度極大。自20世紀30年代以來,全球的科研工作者圍繞著6Li/7Li的分離問題,開展了大量的研究工作,發展了系列的鋰同位素分離方法,如鋰汞齊法[3]、離子交換色層法[4]、溶劑萃取法[5-6]、激光分離法[7]、電沉積法[8]等。其中鋰汞齊法是目前唯一實現工業化的方法,其同位素分離因子為1.020~1.056。雖然鋰汞齊法分離因子高,相間傳質速度快,但生產過程中需要大量的劇毒物質——汞。據報道[1],截至2012年,美國已經使用10886 t的汞用于6Li/7Li的分離,而汞的泄漏可能帶來災難性的環境和安全問題,美國國家環境保護局(EPA)已禁止采用鋰汞齊法生產鋰同位素。因此開發一種安全、高效的鋰同位素分離技術成為研究熱點。
化學交換法分離鋰同位素工藝簡單、連續操作性強,受到研究者的廣泛關注。自Pedersen[9]發現了冠醚以及Lehn等[10]發現穴醚化合物以來,冠醚、穴醚及其衍生物用于鋰同位素分離得到了廣泛的研究[5-7]。由于其特殊的空穴結構和尺寸效應,其對鋰同位素的分離因子可以和汞齊法相媲美,是目前研究和報道最多的鋰同位素分離體系,其對于鋰同位素的分離是基于6Li、7Li在兩相中形成鋰配合物的離子交換反應。我國蘭州大學、原子能研究院、上海有機所開展了不同結構的冠醚對鋰同位素分離的研究,證實鋰同位素效應與兩相化合物種的鍵強差有關,冠醚空穴的大小、結構及邊環上的取代基、有機溶劑的類型、鋰鹽陰離子等影響鋰同位素在兩相中的分配[11-13]。日本Nishizawa課題組[14-16]開展了大量冠醚萃取分離鋰同位素的研究,綜合6Li/7Li的分離因子和鋰離子的分配系數,確定了苯并15-冠-5-LiI/H2O是最佳的鋰同位素分離體系。韓國Kim課題組[4,17-18]考察了大環聚醚固載的離子交換樹脂制成的色譜柱對鋰同位素的分離效應,獲得了較高的6Li/7Li分離因子。以上代表性的結果如表1所示。近年來,隨著新技術、新介質和新材料的層出不窮,冠醚分離鋰同位素的離子交換體系又取得了長足的進展。目前關于鋰同位素分離的綜述多是圍繞傳統鋰同位素分離方法展開的[19-21],缺少對近年來新材料和新介質的總結,且少有關于同位素分離理論的研究報道。為了進一步推進鋰同位素分離技術的發展,有必要總結近年來鋰同位素分離取得的新發展和新突破。
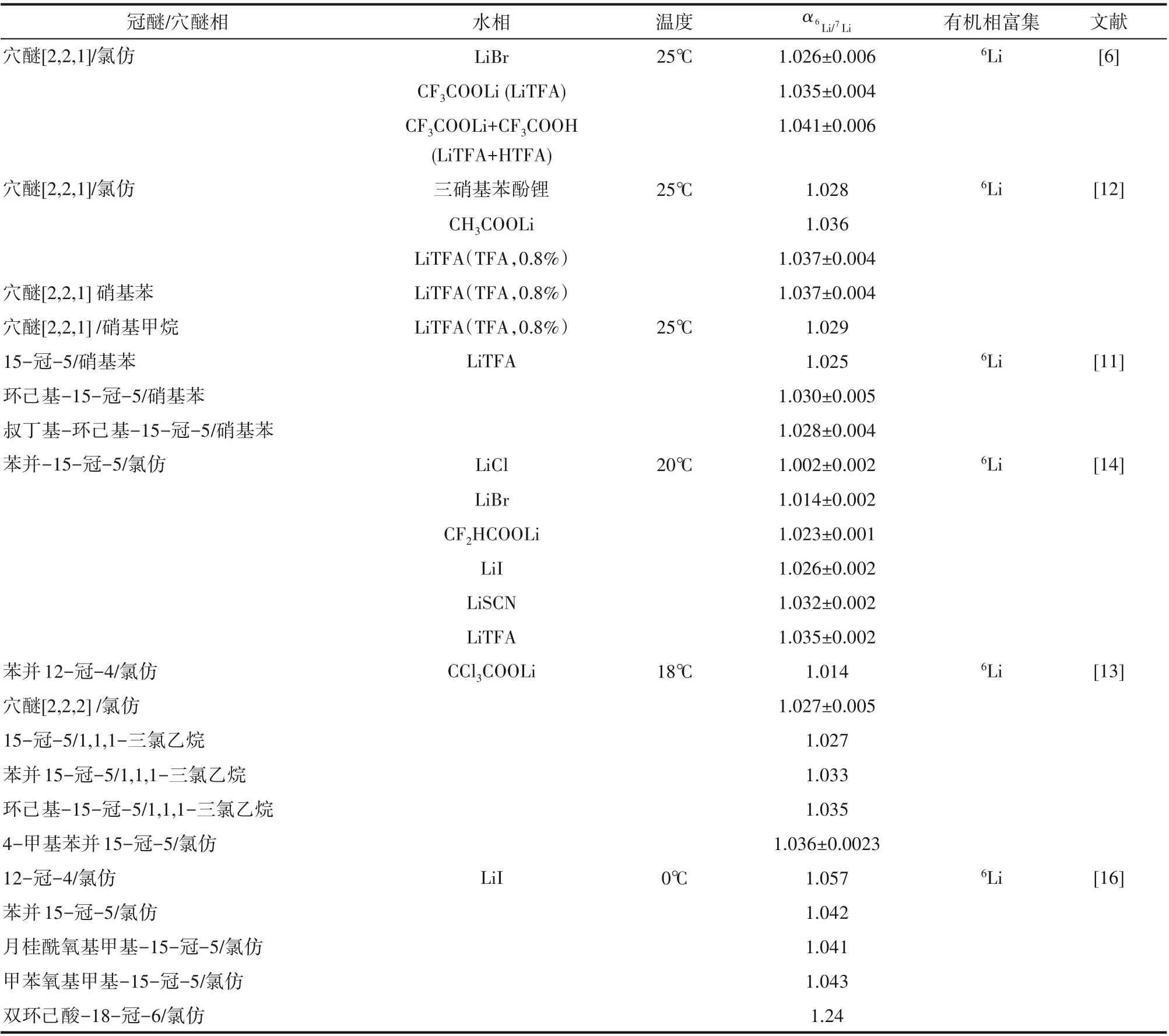
表1 冠醚/穴醚離子交換體系分離鋰同位素Table 1 Separation of lithium isotopes by crown ether/cryptand ion exchange systems
本文通過Web of Science工具對1966—2020年發表的鋰同位素分離的相關文章進行分析。首先對現有的化學交換法分離鋰同位素的理論進行介紹。針對目前冠醚分離鋰同位素存在的鋰離子分配系數低,不利于鋰同位素多級分離和富集的問題,總結了近年來在強化冠醚和鋰離子絡合方面的研究進展,重點綜述了冠醚膜材料、冠醚硅材料、離子液體強化冠醚分離鋰同位素的體系。最后對鋰同位素分離未來的發展方向進行了展望,以期為國內外相關的科研工作者提供參考和指導。
1 文獻統計分析
以“lithium isotope separation”為關鍵詞,采用Web of Science工具對1966—2020年期間發表的489篇論文進行統計分析。如圖1(a)所示,在2000年、2018年左右分別出現鋰同位素分離研究的熱潮。前一個關于鋰同位素分離的研究探索主要集中于離子交換色層法,離子交換劑包括有機離子交換劑和無機離子交換劑,有機離子交換劑主要是冠醚螯合樹脂,無機離子交換劑包括錳氧化物離子交換劑、鈦基離子交換劑、鋯基離子交換劑等,此外還有激光分離法、石墨插層法、電化學法、氣相沉積法等。隨著膜分離技術、材料科學的發展以及離子液體等新介質的出現,鋰同位素分離出現了新的研究熱潮,這一階段開展的鋰同位素分離研究主要集中在冠醚固載聚合物如膜材料、硅材料和纖維材料以及離子液體和冠醚組成的分離體系。如圖1(b)所示,從論文數量上來看,鋰同位素分離研究主要集中在日美中三國,說明這三國的科研人員投入了相對較大的精力,具有較高的論文產量。
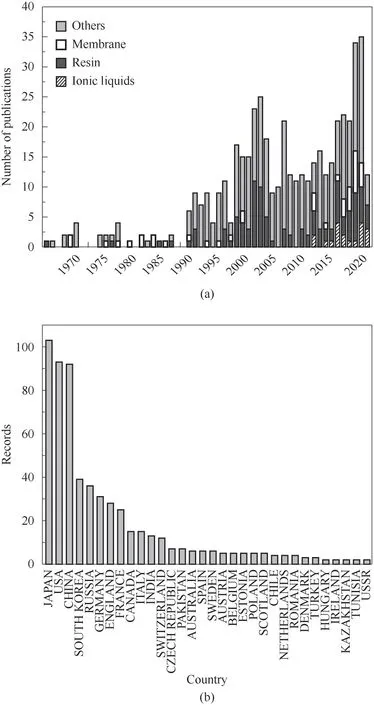
圖1 Web of Science中“lithium isotope separation”為主題的論文分布Fig.1 Publications on Web of Science about the separation of lithiumisotopes
2 鋰同位素分離理論
2.1 Urey模型
20世紀40年代,美國的Urey[22]和Biegleisen等[23]最早提出利用量子化學的觀點以同位素分子簡諧振動頻率來估算同位素交換平衡常數(K)的方法(Urey模型或Bigeleisen-Mayer公式)。該模型是穩定同位素地球化學的基石,認為同位素分餾的本質是同位素間質量上的差異,質量上的不同決定了同位素交換反應達到平衡時生成物和反應物的能量差別。對于同位素交換反應:

式中,X、X'分別代表輕、重質同位素,同位素分離因子α與同位素交換平衡常數K的關系為α=K1/n,其中n為交換的原子數。對于鋰同位素交換反應,即α=K。同位素交換平衡常數K又可以表示為:
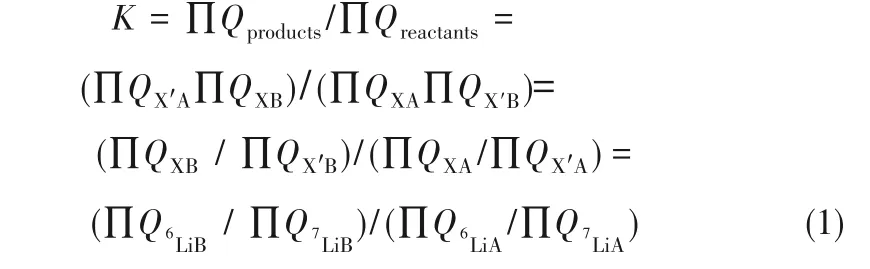
式中,Q為分子配分函數,求出了同位素交換反應反應物和生成物分子配分函數的比值,就可得到α。根據統計力學,粒子的配分函數可表示為:
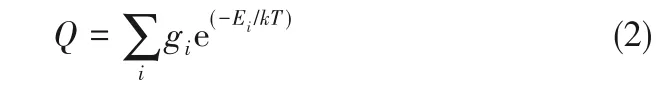
式中,Ei是粒子的能級,gi是對應于能級Ei的簡并度,T為熱力學溫度,k為Boltzmann常數。其中的能量項包括核自旋運動能Enuc、電子能Telec、平動能Etran、轉動能Erot、振動能Evib。因此分子的配分函數可以表示為各種運動形式配分函數的乘積:
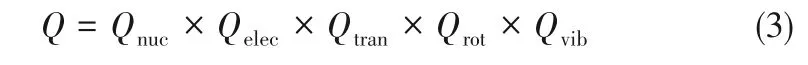
其中分子振動能配分函數Qvib在同位素在兩相的分配中起主要作用,可以表示為:
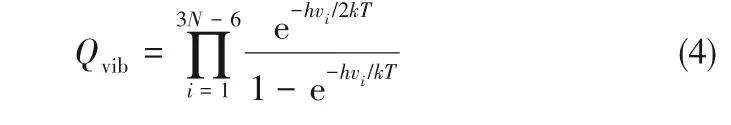
式中,h是Planck常數,為6.626×10-34J·s;k是Boltzmann常數,為1.38×10-23J/K;T是熱力學溫度,K;N是鋰配合物的原子個數(對于非線性分子來說,i=3N-6);vi為振動頻率,Hz。Urey模型引入了約化配分函數比率的概念,用β表示,6Li/7Li同位素分離因子α就可表示為兩相中鋰配合物約化配分函數比率的比值:
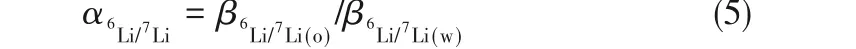
式中,下角標o和w分別代表有機相和水相。
根據Teller-Redlich乘法定則和對分子轉動的近似處理,β可簡化表示為:

因此,確定了兩相中同位素配合物分子的簡諧振動頻率,就可以計算離子交換反應的鋰同位素的分離因子,Urey模型的更多的信息和公式推導可參考文獻[23-24]。
該模型主要用來預測地球化學領域同位素的分餾現象。由于早期缺乏對液相和固相體系分子內部信息的了解,該模型最先用于預測氣體同位素分子的分餾機制。隨著溶液化學、量子化學計算、晶體測試技術等的發展,Urey模型在地球化學領域研究穩定同位素分餾有了很多應用[25-31]。Liu等[27-29]采用Urey模型和量子化學方法成功地預測了各種地質體系Ge、Se、Si同位素的分餾平衡。Anbar等[26]和Jarzecki等[30]基于Urey模型研究了水溶液中[Fe(H2O)6]2+和[Fe(H2O)6]3+之間的Fe的同位素分餾。關于6Li/7Li的同位素分餾也有報道[32-34],Jahn等[32]采用Urey模型和從頭算法,預測了6Li/7Li在水溶液或在含鋰礦物及流體之間的平衡分餾系數,但研究主要基于高壓和高溫的地球和地幔條件下。Cui等[35]采用Urey模型和量子化學計算成功預測了溶劑萃取體系冠醚-有機相/鋰鹽-水相的鋰同位素效應,并從化學鍵、振動頻率等微觀尺度揭示了化學交換過程中6Li/7Li分離的影響因素及其機制。結果表明冠醚的空腔尺寸是影響鋰同位素分離的主要因素,冠醚屏蔽水相中與Li+絡合的水分子能力越強,6Li/7Li的分離因子越大。通過削弱鋰-冠醚絡合物的Li—O鍵有助于提高6Li/7Li分離因子,例如,使用強極性有機溶劑、選擇軟堿陰離子的鋰鹽開展同位素的分離研究。
2.2 鍵的力常數理論
1977年,Betts等[36]根據萃取過程中同位素交換反應平衡常數K與配分函數相關,借助一級量子修正和忽略矩陣元素G、F推導出K的簡單表達式:
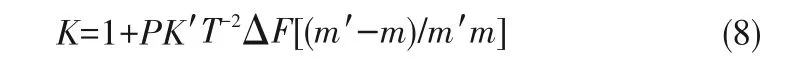
式中,P為金屬離子的配位數,K'=(h/2πk)2/24,T為熱力學溫度,m'和m分別為重同位素和輕同位素的質量,ΔF是有機相和水相中金屬-配體鍵的力常數之差。從式(8)可以得出同位素分離因子取決于兩相絡合物中金屬-配體化學鍵的力常數之差(∣ΔF∣),∣ΔF∣越大,K越大;因此,配體的選擇對于同位素的分離至關重要。另外,金屬離子的配位數(P)越大,K越大;其中重同位素富集在強鍵相,輕同位素富集在弱鍵相。
對于鋰同位素萃取分離的離子交換體系,鋰離子在水相中以水合形態[Li(H2O)4]+存在(Li—O的鍵長約為1.9~2.0?[35],1?=0.1 nm),鋰同位素的分離主要與鋰在有機相中的絡合形態及鍵合強度有關。當有機相為冠醚螯合體系時,由于鋰離子和冠醚的結合是離子-偶極作用(Li與冠醚O原子形成的化學鍵鍵長為2.1~2.7?)[圖2(a)、(b)],與水相相比,屬于弱鍵相,易富集6Li(表1)。對于酸性螯合劑如偶氮萘酚/羥基喹啉/雙酮類和中性配體或離子液體組成的協萃體系,Li+與配位O或N原子形成較強的配位鍵(Li—O/N化學鍵的鍵長在1.9~2.1?)[圖2(c)、(d)],與水相相比,屬于強鍵相,易富集7Li(表2)。

表2 酸性螯合/中性配體或酸性螯合/胺類萃取劑協萃體系的鋰同位素效應Table 2 Lithium isotopic effect of acid chelating agent/neutral ligand or acid chelating agent/amine extractant co-extraction systems
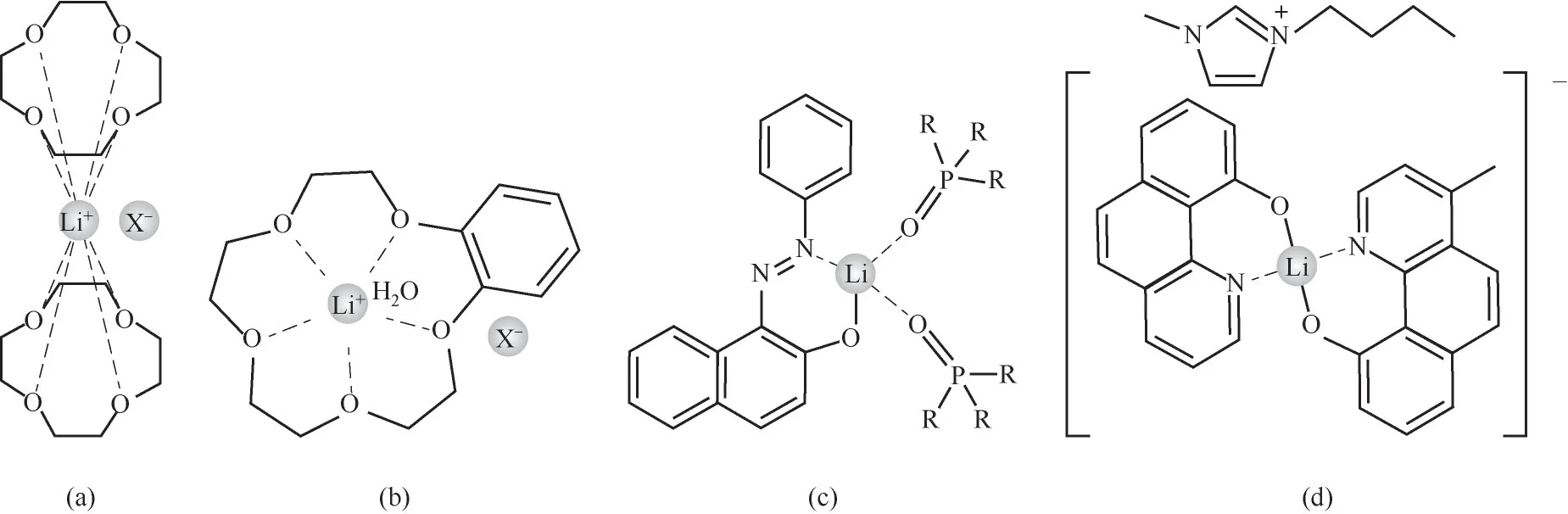
圖2 冠醚和酸性螯合體系與Li+形成的配合物結構:(a)12-冠-4與Li+形成的配合物(X為I時,Li—O的平均鍵長為2.43?)[35];(b)苯并-15-冠-5與Li+形成的配合物(X為I時,Li—O的平均鍵長為2.24?)[35];(c)1-苯基偶氮-2-萘酚/TOPO與Li+形成的配合物[37](R=C8H17);(d)4-甲基-10羥基苯并喹啉/1-甲基3-甲基咪唑氯化物與Li+形成的配合物[38]Fig.2 The complex structures of crown ether or acidic chelating agent with Li+:(a)the complex of 12-crown-4 with Li+(the average bond length of Li—Ois 2.43?when X is I)[35];(b)the complex of benzo-15-crown-5 with Li+(the average bond length of Li—Ois 2.24?when X is I)[35];(c)the complex of 1-phenylazo-2-naphthol/TOPOwith Li+(R=C8H17)[37];(d)the complex of 4-methyl-10 hydroxybenzene quinoline/1-methyl 3-methylimidazole chloride with Li+[38]
2.3 熱力學理論
嚴峰等[19]從熱力學角度解釋了冠醚分離鋰同位素的機制。他們認為,同位素分離效應的本質是冠醚對6Li、7Li絡合自由能變的差異引起的,由式(9)、式(10)兩個平衡式獲得:
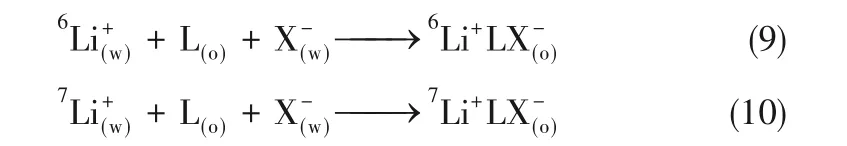
式中,L代表冠醚配體,X-是鋰鹽陰離子。式(9)與式(10)的自由能變差異越大,6Li/7Li分離因子越大。冠醚絡合Li+的凈自由能變ΔG?由體系焓變ΔH?和熵變ΔS?共同組成,具體表示為冠醚-鋰絡合的焓與游離溶劑分子增加所獲得的熵的和,減去冠醚與鋰離子去溶劑化以及冠醚構象重排引起焓的損耗。因此,任何影響該過程的自由能變的因素都影響到鋰同位素效應,包括冠醚的分子結構、給體原子的種類、溶劑類型、反配陰離子等。在各種過程的互相影響下最終形成冠醚在某一體系的鋰同位素效應。
理論上,式(9)和式(10)中的ΔG?、ΔH?、ΔS?需要純6Li、7Li與冠醚的熱力學實驗,難以獲得。所以在鋰同位素交換體系中,ΔG?、ΔH?、ΔS?主要通過式(11)得到:
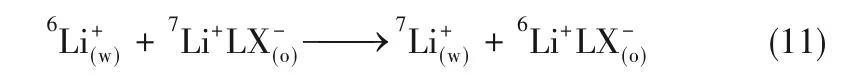
Nishizawa等[16]研究了不同空穴尺寸冠醚的鋰同位素效應,從熱力學角度闡述了冠醚空穴尺寸對6Li/7Li分離因子的影響。表3為冠醚-氯仿/LiIH2O離子交換體系在0℃的熱力學函數和分離因子,發現隨著冠醚空穴尺寸的增大,同位素交換反應的焓變、熵變絕對值降低,對應的α6Li/7Li也降低,推測這是由于冠醚尺寸的增大增加冠醚構象重排時焓損耗[19],自由能變降低,從而降低分離因子。
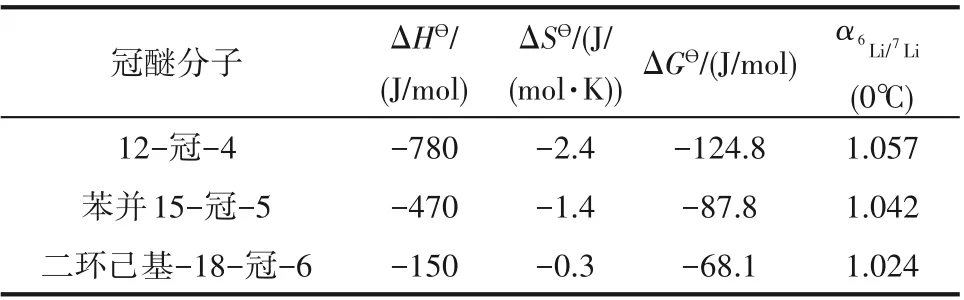
表3 不同尺寸冠醚分離鋰同位素的熱力學函數[19]Table 3 Thermodynamic analysisof 6Li/7Liseparation by crown ethers with different sizes[19]
3 鋰同位素分離研究新進展
由于冠醚顯著的鋰同位素效應,近年來,鋰同位素分離的新材料和新體系的研究基本上都是圍繞冠醚展開的。Li+分配系數和6Li/7Li分離因子對于鋰同位素分離來說是兩個重要的指標,但兩者是不同過程的結果,Li+分配系數對應于溶劑萃取或者吸附過程Li+在兩相分配達到平衡的結果,而6Li/7Li分離是6Li、7Li在兩相中鋰配合物之間離子交換達到平衡的結果。如何平衡兩者之間的關系是鋰同位素分離的一個關鍵科學問題。理想的化學交換法分離鋰同位素既要求有較高的Li+分配系數,又有適宜的鋰同位素分離因子。單從分離因子上看,冠醚是最有前途實現工業化鋰同位素分離的離子交換劑,但Li+在冠醚和水溶液兩相的分配系數低,不利于鋰同位素的多級分離和富集,這是限制同位素分離工業化應用的主要原因。除此之外,溶劑萃取過程中冠醚小分子易流失也是分離的一個弊端。為了解決上述問題,科研工作者做了一系列的研究探索。
3.1 離子液體強化冠醚分離鋰同位素
離子液體(ionic liquids)作為一種綠色的反應和分離介質,有可忽略的蒸氣壓、不易揮發、不易燃燒、熱穩定性好等優點。離子液體作為稀釋劑取代溶劑萃取過程中揮發性有機溶劑以及離子液體作為分離介質參與分離過程已有很多應用[42-44]。研究表明,在有中性配體如磷酸三丁酯(TBP)、三烷基氧化膦(Cyanex 923)、三辛基氧化膦(TOPO)、冠醚等存在的條件下,[PF6]-和[NTf2]-基疏水性離子液體可顯著提高中性配體對金屬離子的分配系數[45-48],萃取分離機制為陽離子交換。以[OHEmim][NTf2]強化Cyanex 923從鋰鹽水溶液中萃取Li+為例[49]:
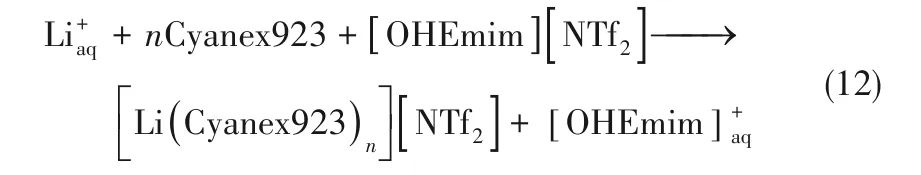
Xiao等[50-53]在研究離子液體強化冠醚提鋰和分離鋰同位素方面開展了大量研究,考察了不同鋰鹽類型、咪唑基離子液體烷基鏈長度、水相pH、不同空穴的冠醚等對Li+分配系數和6Li/7Li分離的影響。結果表明,降低烷基鏈的長度有利于Li+分配系數的提高,在最佳pH=6的條件下,離子液體的加入可使Li+在相間的分配系數從10-3~10-5[14-15]提高到10-2~10-1數量級[50],形成了(LiLn)+Y-復合物(L表示冠醚,Y表示離子液體陰離子),同時也在一定程度上改善了鋰同位素的分離因子[52],尤其對于陰離子為硬堿的氯化鋰鹽體系,在[C6mim][PF6]存在的條件下,20℃時B15C5對LiCl-H2O體系中6Li/7Li的分離因子為1.032[50]。他們認為,陰離子的核外電子云密度越低,鋰同位素的分離效果也越好,對不同的鋰鹽體系,6Li/7Li單級分離因子從大到小的順序為:CF3COOLi>LiI>LiBr>LiCl,符合軟硬酸堿理論。Cu等[54-56]也嘗試將陰離子為[PF6]-、[BF4]-和[NTf2]-的[C8mim]基離子液體作為溶劑強化冠醚體系提鋰以及6Li/7Li的分離。為減少冠醚的水溶性,他們設計合成了含有萘環的2,2'-聯萘二基-17-冠-5醚(BN-17C5),發現[C8mim][NTf2]對冠醚提鋰的強化作用較好,25℃時BN-17C5-[C8mim][NTf2]從CF3COOLi-H2O溶液中獲得1.046的6Li/7Li分離因子,Li+的分配系數為0.06[54]。為了解決由于離子液體高黏度度帶來的傳質速率慢的問題,他們將離子液體和冠醚通過溶膠凝膠法固定在介孔硅材料[55]或者浸漬法固載在樹脂材料[56]。
離子液體和冠醚組成的離子交換體系和單純冠醚相比,可提高Li+的分配系數。同時在萃合物中引入離子液體的陰離子[PF6]-或[NTf2]-作為平衡電荷,由于較大的陰離子體積和較小的電子云密度,削弱了與(LiLn)+之間的靜電作用,增強了水相和冠醚相Li—O鍵的力常數之差。基于2.2節鍵的力常數理論,有利于同位素效應的改善,但離子液體陽離子的損失是該方法的弊端。
3.2 冠醚固載材料分離鋰同位素
鑒于冠醚在溶劑萃取過程中小分子易流失的問題,研究者將冠醚通過化學接枝的方法固載于介孔二氧化硅、膜材料以及其他高分子聚合物上,采用固-液萃取法分離鋰同位素。該方法使得冠醚不需要溶解在溶劑中,鋰鹽的溶劑也不再局限于水,可以溶于比水極性低的有機溶劑中,如乙腈(C2H3N)、丙酮(C3H6O)等,避免了Li+的水合作用帶來的負面影響,使Li+去溶劑化過程焓損耗減少,有利于Li+和冠醚的結合[57-58]。另一方面,也避免了冠醚和Li+相互作用過程中配位水分子的引入,這對基于冠醚體系的鋰同位素分離是有利的。
3.2.1 冠醚固載硅材料 過去20年里,有序介孔二氧化硅(OMS)為功能化納米材料的研究帶來了巨大活力,其高比表面積、均勻的通道結構、可調的孔道大小和形狀為引入特定的有機配體創造了條件,功能化后的OMS可以有效吸附和分離金屬離子[59]。Ye等[60]首先合成一系列含有不同取代基(—NH2、—NO2、—C4H9)的B15C5衍生物,在液-液萃取體系中考察了B15C5及其衍生物的鋰同位素效應,結果表明供電子效應強的—NH2取代的B15C5具有較好的和Li+結合以及6Li/7Li分離性能,—C4H9取代的B15C5次之,—NO2取代的B15C5和Li+結合以及6Li/7Li分離性能最差。他們采用導入接枝(postgraft)法[60]和連續活化劑再生引發劑引發的原子轉移自由基聚合(ICAR ATRP)技術[61]將NH2-B15C5固定在SBA-15硅基介孔材料上(圖3),研究其在固-液萃取條件下的提鋰效果和鋰同位素分離效應。結果表明,在具有較軟鋰鹽陰離子的體系以及將鋰鹽溶于較低介電常數的溶劑中時,可獲得較大的同位素效應。熱力學研究表明,上述B15C5衍生物和Li+的結合以及鋰同位素的離子交換反應均為放熱反應,降低溫度有利于提鋰和6Li/7Li的分離,該材料在288.15 K、CF3COOLi-C2H3N體系中獲得最高6Li/7Li分離因子1.049[60],此時Li+的吸附容量約為4.0~5.0 mg/g。
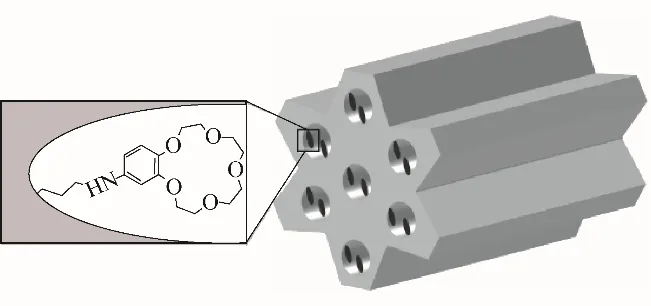
圖3 氨基-苯并-15-冠-5接枝SBA-15材料的結構[60]Fig.3 The illustration of amino-benzo-15-crown-5 grafted SBA-15 material[60]
3.2.2 冠醚固載聚合物材料 將大環聚醚通過化學反應接枝在聚合物材料上也有大量的報道。嚴峰課題組[19,62-69]在冠醚功能化的膜材料分離鋰同位素方面開展了大量研究,通過縮醛化[67]、親核取代[69]等合成方法將甲酰基、胺基等功能基團取代的B15C5或氮雜15冠5醚接枝在聚乙烯醇(PVA)、聚砜(PSF)等聚合物材料上(圖4),考察了其在固-液萃取條件下的鋰同位素效應,研究了冠醚固載量、鋰鹽陰離子、反應溫度、鋰鹽溶劑等對鋰同位素分離的影響。結果表明,冠醚的固載量對Li+的分配系數和鋰同位素分離因子影響較大,提高負載量有利于Li+的分配系數和同位素分離因子的提高,具有較軟陰離子的鋰鹽以及采用較低介電常數的溶劑制備鋰鹽相時可獲得較高的Li+分配系數和鋰同位素分離因子。如對于4'-甲酰基苯并15-冠-5接枝聚乙烯醇(PVA-g-FB15C5)和無紡布形成的復合材料從LiI的水溶液中提鋰和分離鋰同位素,當冠醚的接枝量從0.76 mmol/g提高到2.13 mmol/g時,Li+的分配系數可從136 ml/g提高到175 ml/g,鋰同位素的分離因子從1.013提高到了1.065,大于4'-甲酰基苯并15-冠-5和鋰鹽-水溶液組成的液-液萃取體系(α=1.022)以及PVA-g-FB15C5和鋰鹽-水溶液組成的固-液萃取體系(α=1.046)的鋰同位素效應,Yan等[62]認為,同位素分離因子的顯著改善與復合材料的孔結構有關。
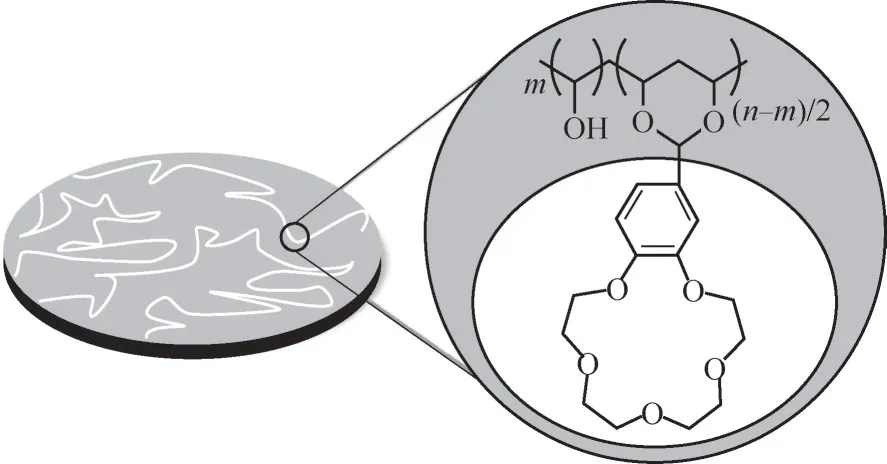
圖4 PVA-g-FB15C5的結構[62]Fig.4 The structure diagram of PVA-g-FB15C5[62]
也有研究者將B15C5通過化學反應接枝到纖維材料上用于鋰同位素分離。Zhai等[58]通過γ射線輻射誘導乳液接枝技術制備了具有4-氨基苯并15-冠-5活性單元的纖維素微球(MCM-g-AB15C5)。Ye等[57]通過ICAR-ATRP技術將—NH2取代的B15C5固定在玻璃纖維上并研究了其對Li+的吸附和鋰同位素的分離行為。盡管目前有大量的冠醚固載材料分離鋰同位素的報道,但受到冠醚接枝量的限制,普遍存在吸附容量低的問題。
3.3 含氟溶劑萃取體系
胡金波課題組[41,70-71]在鋰同位素分離方面做了大量的研究工作,建立了鋰同位素多級富集分離的成套裝置,國際上首次實現利用有機萃取法富集分離得到豐度為99.992%的7Li樣品。他們先后公開了可有效富集7Li的萃取劑,分別是含氟的偶氮萘酚萃取劑[70][圖5(a)]和苯并喹啉類萃取劑[41][圖5(b)]。其中含氟萃取劑萘環上的—OH基團具有較高的質子酸性,在堿性條件下可以失去質子,在偶氮基團和季銨鹽協萃劑的共同作用下,可與Li+形成穩定的螯合物((SF-Li-SF)-L+,SF為去質子的含氟萃取劑,L為協萃劑),具有較高的萃取率和鋰同位素分離因子。同時含氟基團的存在也改變了偶氮基團中氮原子對鋰的配位性能,產生顯著的同位素分離效應,反萃后,萃取劑和協萃劑可循環使用。在5-氟-9-甲氧基-10-羥基苯并喹啉/N263/二氯苯/LiCl體系中,Li+的分配系數為1.5,7Li/6Li的最大單級分離因子為1.028,采用(NH4)2SO4作為反萃劑,三次反萃率達99%,實現了鋰同位素多級分離和富集。該課題組還公開了一類含氟的冠醚萃取劑[圖5(c)]用于鋰同位素分離[71],由于含氟基團的引入,使該類冠醚疏水性強,分相快、效率高,鋰同位素的單級分離因子為1.033~1.041,其中冠醚相富集6Li。由于含氟基團的吸電子效應,削弱了鋰-冠醚配合物中的Li—O鍵,根據Urey模型和鍵的力常數理論,有利于6Li/7Li分離。
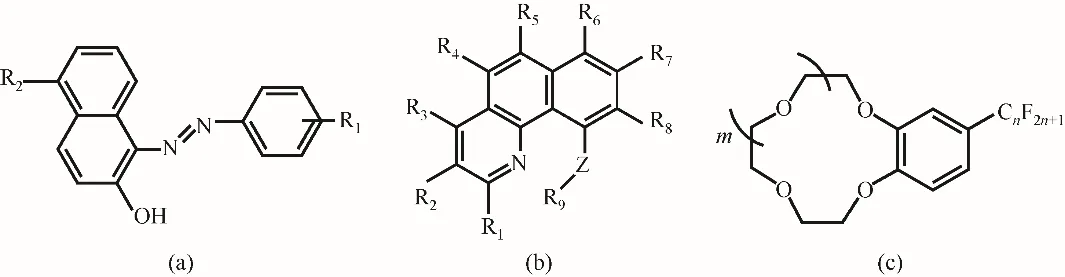
圖5 含氟萃取劑結構示意圖:(a)偶氮萘酚萃取劑[70](R1為氫、溴、氯、氟或C1~6氟代烷烴,且至少有一個為氟或C1~6氟代烷烴;R2為氫、鹵素、硝基、氰基或C1~6氟代烷烴);(b)苯并喹啉萃取劑[41](Z為氧、硫或氮原子;R9為氫、磺酰基;R1~8為氫、鹵素、苯基等);(c)含氟冠醚萃取劑[71]Fig.5 The structures of the fluorine-containingextractants:(a)azo-naphthol extractant[70](where R1 is hydrogen,bromine,chlorine,fluorine or C1—6 fluoroalkane,and at least one is fluorine or C1—6 fluoroalkane;R2 is Hydrogen,halogen,nitro,cyano or C1—6 fluoroalkane);(b)benzoquinoline extractant[41](Zis oxygen,sulfur or nitrogen atom;R9 is hydrogen,sulfonyl group;R1—8 are hydrogen,halogen or phenyl,etc.);(c)fluorine-containing crown ether[71]
綜上所述,無論是溶劑化萃取體系還是固-液萃取體系,冠醚是目前研究和報道最多的鋰同位素分離體系。但冠醚作為活性單元,普遍存在Li+的分配系數低的問題,且Li+的分配系數和6Li/7Li的分離因子受鋰鹽陰離子的影響較大,只有陰離子半徑大、變形性強的陰離子如CF3COO-、SCN-、I-等存在時才可獲得較高的Li+分配系數,冠醚與Li+的結合一般服從Hofmeister序列;對于廣泛存在的親水性的氯鹽和硫酸鹽體系,冠醚和Li+的結合性能很差,且6Li/7Li的分離因子也不高。這主要由Li+的電子結構決定,Li+具有1s2的惰性電子結構,無d軌道,化學性質穩定;離子勢小,配位能力差;通過單一的相互作用很難將Li+從高價離子共存的體系中選擇性分離出來,提取難度很大。針對冠醚和Li+結合性能差,研究者通過構建質子化的冠醚或者在大環聚醚體系中引入氫鍵供體、Lewis酸等陰離子識別位點可以顯著改善Li+的分配系數,這些研究有望為鋰同位素分離新體系和新方法的建立提供思路。
4 新型冠醚離子對體系
4.1 質子化冠醚
研究表明,相比中性冠醚,在堿性條件下,側鏈帶有質子化基團(—COOH、—PO3OH等)的冠醚分子(圖6)容易脫去質子,生成共軛堿,與Li+絡合時可形成更加穩定的配合物。Torrejos等[72]合成了側臂具有羧酸基團的二苯并14-冠-4(DB14C4-C18-COOH),以離子液體三己基十四烷基雙(三氟甲基磺酰基)酰胺(CYPHOSIL 109)為稀釋劑,在pH=12.8、冠醚濃度為0.025 mol/L時,DB14C4-C18-COOH對Li+的分配系數為6.6,大于DB14C4-C18和DB14C4為萃取劑時Li+的分配系數(<0.21)。將—COOH基團固定在接枝了冠醚環的多壁碳納米管上也觀察到相同的趨勢[73]。Habata等[74]將可電離的磷酸官能團(—PO3OH)與冠醚結合,在pH=11時,含磷酸官能團的DB14C4與羥甲基二苯并-14冠-4相比,Li+的萃取率提高了約270倍,且具有較高的Li+/Na+和Li+/K+選擇性。
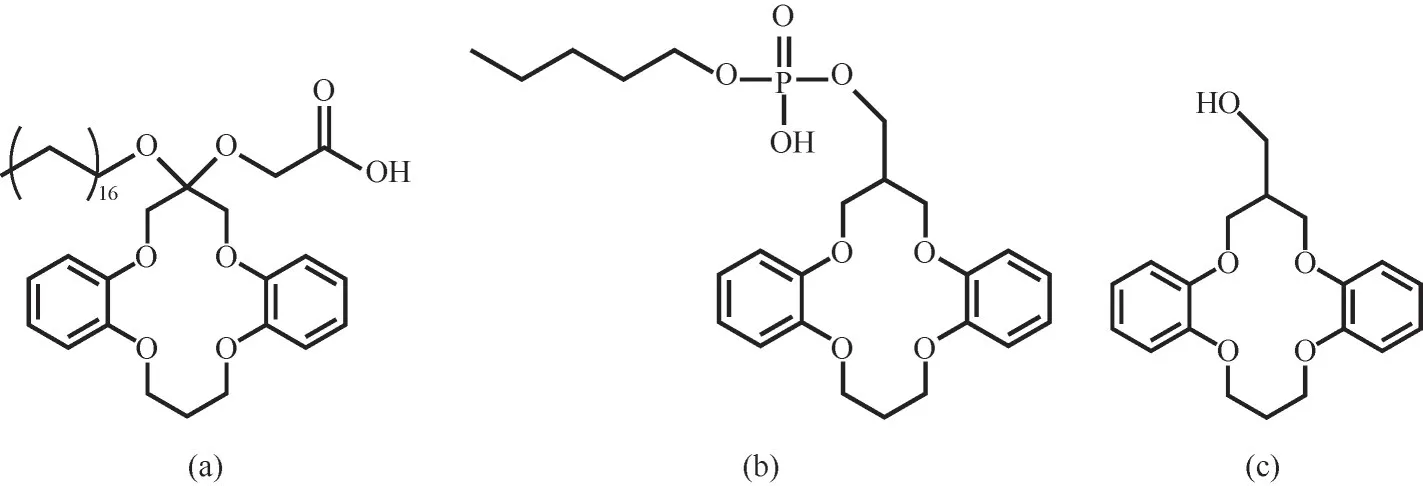
圖6 側鏈帶有質子化基團(—COOH、—PO3OH等)的冠醚分子的結構:(a)DB14C4-C18-COOH[72];(b)含烷基磷酸官能團的二苯并-14冠-4[74];(c)羥甲基二苯并-14冠-4[74]Fig.6 The structure of crown ether molecules with protonated groups(—COOH,—PO3OH,etc.):(a)DB14C4-C18-COOH[72];(b)dibenzo-14-crown-4 with alkyl phosphate group[74];(c)hydroxymethyl dibenzo-14 crown-4[74]
4.2 雙功能離子對受體
在大環化合物結構中引入脲鍵,形成雙功能離子對受體。由于脲鍵(—NH—CO—,—NH—CS—)可以與F-、Cl-、Br-、NO3-等陰離子形成氫鍵,可用于陰離子的識別,冠醚作為陽離子的識別位點,通過控制冠醚環的大小能選擇性地結合Li+。因此,在大環化合物上引入脲鍵作為氫鍵供體可實現鋰鹽分子的高效提取和特異性識別。如Smith等[75]報道了一個可用于固-液萃取分離過程選擇性提取鋰鹽分子的雙功能配體(圖7)。通過單晶衍射儀測試配合物的結構發現,LiCl和LiBr以離子對的形式進入配體,水分子也參與配位反應;核磁表征顯示,在LiCl/NaCl/KCl-CDCl3三種固體鹽的混合體系中,該雙功能配體對不同鹽的絡合比例為94∶4∶2(摩爾比),表明該配體對Li+有明顯的選擇性。
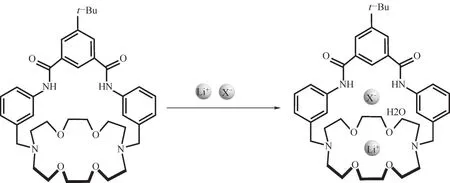
圖7 雙功能配體與Li+的配位反應[75]Fig.7 The coordination reaction of a ditopic salt receptor with lithiumion[75]
在大環化合物中引入金屬作為Lewis酸性位點,形成雙功能離子對受體。Lewis酸位點作為電子對接受體可用于陰離子的識別,通過和冠醚環的協同作用,能選擇性地結合鋰鹽離子對。如Wendji等[76]合成了含有Lewis酸性基團的二茂鐵13-冠-4醚離子對接受體,其和LiCl能形成穩定的配合物,其中冠醚部分選擇性的絡合Li+,Sn起到選擇識別Cl-的作用;通過制備單晶,得到了含Sn的二茂鐵13-冠-4醚與LiCl形成配合物的分子結構,結果表明,LiCl的Li+和Cl-分別和冠醚以及路易斯酸性中心Sn絡合,Li-Cl之間的距離為2.329?(圖8)。

圖8 含Sn的二茂鐵冠醚分子與LiCl形成離子對[76]Fig.8 The complexation of tin-containing crown ether with LiCl[76]
5 結論與展望
6Li、7Li是核能發展重要的原材料,其分離和富集已列為國家科技發展的重大專項。我國從20世紀80年代研究鋰同位素分離以來,已經取得了顯著的進展和突破,其中化學交換反應分離鋰同位素是一個主要的研究方向。溶劑萃取法和吸附法是目前文獻報道最多的鋰同位素分離方法,冠醚是主要的活性單元,與已工業化的鋰汞齊法相比,技術尚未成熟,仍處于實驗室研究階段,但不存在汞泄漏的風險。溶劑萃取法傳質速率快、可連續操作性強,但萃取過程中冠醚小分子易流失是該方法的弊端。將冠醚共價接枝到固相材料雖然可以克服冠醚小分子流失的問題,但由于受到冠醚接枝量的限制,普遍存在吸附容量低的問題,且固-液兩相傳質慢,達到平衡時間長。總之,不管是溶劑萃取法還是吸附法,冠醚作為活性單元,Li+分配系數低,不利于鋰同位素的多級分離和富集,這是限制冠醚用于鋰同位素分離工業化的主要原因。
目前的研究多側重于鋰同位素分離效果的研究,對鋰同位素分離的微觀反應機制研究較少,尚未從電荷、鋰同位素作用的差異、吸附材料孔結構、納微環境、孔道限域效應等微觀尺度揭示分離介質與同位素分離性能之間的構效關系,這在一定程度上也限制了高效分離體系的開發。另外,在分離過程中如何兼顧Li+分配和6Li/7Li分離也是鋰同位素分離亟待解決的關鍵科學問題。因此,鋰同位素分離工業化尚需解決從基礎研究到產業化應用過程中的諸多關鍵科學和技術問題,需要進一步優化萃取劑的結構,研究Li+在分離過程中的動力學和熱力學,開發高效的分離裝置等。鑒于低溫有利于Li+分配及6Li/7Li分離,因此開發低溫的鋰同位素分離技術是今后研究的一大方向。
隨著化石能源的不斷消耗,研發清潔能源已經成為全人類的共識。實現“可控核聚變”雖然困難重重,但從長遠來看,核能必將是人類繼煤、石油和天然氣之后的主要能源,希望通過對鋰同位素分離的基礎理論和工程應用的不斷攻關,推動核能的發展,實現可控核聚變造福人類。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
鐵道通信信號(2020年9期)2020-02-06 09:15:22
科技傳播(2019年22期)2020-01-14 03:06:54
數學大王·趣味邏輯(2019年5期)2019-06-13 20:27:43
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
小學科學(學生版)(2019年5期)2019-05-21 01:00:18
經濟技術協作信息(2018年30期)2018-11-22 06:20:24
新高考·高一物理(2014年1期)2014-09-18 01:26:07
