硅雜原子提升冠醚對鋰離子絡合能力的機理理論研究
2021-06-30 01:28:40梁蘇卓成姬國勛孫新利王波張仕通代星
化工學報 2021年6期
梁蘇卓成,姬國勛,孫新利,王波,張仕通,代星
(1西安高科技研究所,陜西西安710025;2蘇州大學放射醫學與輻射防護國家重點實驗室,放射醫學及交叉學科研究院,江蘇省高校放射醫學協同創新中心,江蘇蘇州215123;3東北電力大學理學院,吉林省吉林市132012)
引 言
冠醚是一類聚醚分子,一般由—CH2—O—CH2—單元構成,擁有環狀柔性骨架結構。根據環內C和O的個數,冠醚被命名為15-冠-5、18-冠-6等,其中第一個數字代表C和O的總個數,第二個數字代表O的個數。冠醚分子整體呈電中性,分子中的O擁有比C和H更高的電負性,因此電子更多地向O聚集,從而能夠通過電荷-偶極以及配位共價鍵等作用絡合金屬離子,具有對堿金屬和堿土金屬的強絡合能力[1-3]。改變冠醚分子環狀骨架的大小、引入N或S等雜原子、增加官能團、連接具有不同功能的側臂能夠調整冠醚對金屬離子的絡合性能,使其具有對特定金屬離子的選擇絡合性[4-6]。引入具有發光特性的官能團,結合冠醚的強絡合能力,研究者獲得了多種具有金屬離子熒光識別能力的冠醚[7-9]。由于冠醚對金屬離子的絡合與熒光識別都以冠醚和金屬離子的相互作用為基礎,因此通過結構設計得到對金屬離子作用能力更強的冠醚一直是研究熱點。
Si與C在元素周期表中屬于同族元素,化學性質具有相似之處,然而相應的Si與C化合物可能表現出截然不同的性質,例如室溫下CO2呈氣態而SiO2為晶體。近年來,含有Si雜原子的類醚分子與金屬離子的相互作用受到了研究者的關注[10-12]。由于Si具有比C更弱的電負性,含有Si雜原子的冠醚中與Si相鄰的O會帶有更多的負電荷,因此可能與金屬離子產生更強的電荷-偶極相互作用與配位共價作用,從而擁有比不含Si的有機醚更強的金屬離子絡合能力。然而研究表明,由于O與鄰近的Si—H、Si—Me反鍵軌道產生的超共軛作用[13]、帶有更多正電荷的Si對金屬離子產生的庫侖互斥作用,以及更大的形變能[14],Si雜冠醚往往表現出比有機冠醚更弱的金屬離子絡合能力[15]。對此,H?nisch等[16-18]基于實驗與密度泛函理論(DFT)計算結果提出了不同的觀點,認為當相鄰的O間隔兩個—SiMe2—單元時,Si雜冠醚能夠比相應的有機冠醚更穩定地絡合金屬離子,例如1,1,2,2-4甲基-1,2-2硅雜12-冠-4擁有比12-冠-4更強的鋰離子絡合能力。盡管H?nisch等對Si雜冠醚與金屬離子的相互作用研究僅停留在配位鍵鍵長、相互作用能和形變能層面,但是足以證明,通過合理引入Si雜原子,可以獲得與金屬離子相互作用能力更強的冠醚材料,從而進一步提升冠醚類金屬離子吸附、熒光識別材料的性能。
目前,Si雜冠醚結構設計所需的理論基礎還不夠成熟,Si雜冠醚的性質及其與金屬離子的相互作用機理仍有待深入理解。因此,本文通過DFT理論計算,詳細地探討了1,1-2甲基-1-硅雜12-冠-4(以下簡稱LSi/C)、1,1,2,2-4甲基-1,2-2硅雜12-冠-4(以下簡稱LSi)、1,1,2,2,4,4,5,5-8甲基-1,2,4,5-4硅雜12-冠-4(以下簡稱LSi-O)、1,1,2,2,7,7,8,8-8甲基-1,2,7,8-4硅雜12-冠-4(以下簡稱LSi-M)及原始12-冠-4(以下簡稱L)與Li+的相互作用,并揭示了Si雜冠醚對Li+絡合增強的機理。考慮到在實際的工程應用中,冠醚與Li+的絡合反應往往在溶劑中進行,而溶劑環境對此類絡合反應的Gibbs自由能變有不可忽略的影響[19]。因此,本文還計算了氣相條件下和二氯甲烷溶劑條件下5種冠醚-Li+絡合反應的Gibbs自由能變,以研究溶劑中5種冠醚對Li+的絡合能力,以及有機溶劑二氯甲烷對絡合反應的影響。
1 模型與計算方法
基于前人的研究基礎,即認為當—SiMe2—SiMe2—單元取代相鄰的O間—CH2—CH2—單元時,雙Si雜冠醚能夠比相應的有機冠醚更穩定地絡合金屬離子。此摻雜方式不會改變冠醚大環原子數,對于12-冠-4來說,摻雜前后均為12元環。因此本工作采取摻雜Si數量為1的LSi/C、摻雜Si數量為2的LSi、摻雜Si數量為4的LSi-O和LSi-M以及原始未摻雜的12-冠-4共5種冠醚作為理論計算模型(圖1),探究5種冠醚對Li+的相互作用。本工作的研究內容同時涉及Si的摻雜數量和摻雜位點對冠醚性質的影響。以—SiMe2—取代—CH2—CH2—的單硅雜冠醚已經被實驗確認其絡合Li+能力不如原始冠醚[15],主要原因理論上被解釋為帶正電的Si與Li+產生庫侖排斥[11],因此該模型不在本文討論范圍。
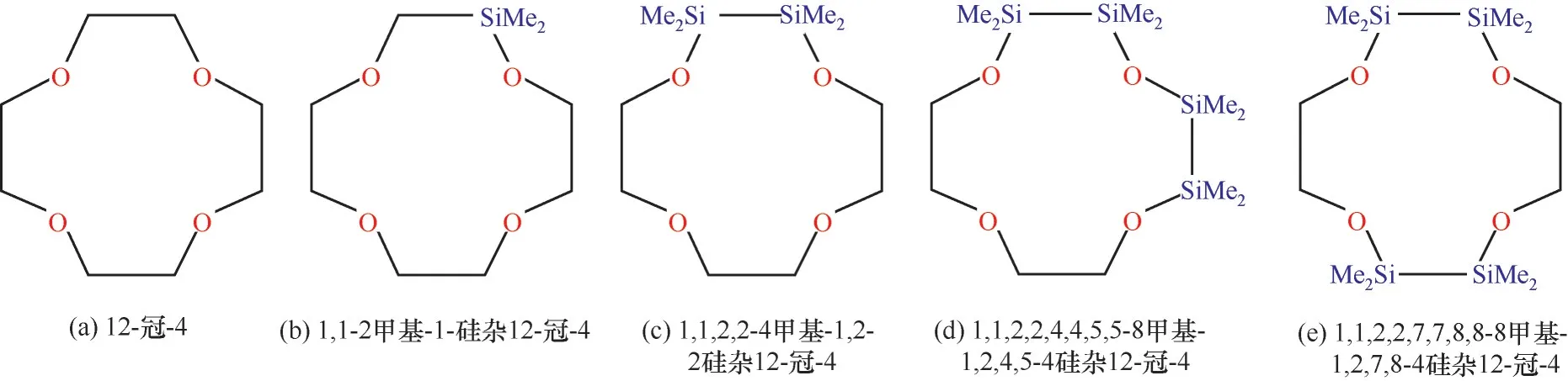
圖1 冠醚的化學結構式Fig.1 The chemical formulas of crown ethers
由于冠醚-金屬離子絡合中的相互作用大量涉及電子層面的機理,高精度量子化學計算適合研究此類問題[20-22]。基于計算精度和效率的同時考慮,本文采用Gaussian 09程序對所有體系執行密度泛函理論計算。
在B3LYP-D3(BJ)/Def2SVP級別[23-28]對體系進行幾何優化。優化過程中沒有對對稱性施加任何限制。在相同理論級別下進行了頻率計算,沒有虛頻以保證得到的結構是勢能面上真正的駐點。首先優化的結構是12-冠-4。而后在所得的穩定12-冠-4結構上摻雜Si并進行優化,得到4種Si雜12-冠-4的穩定結構。最后在冠醚環內增加Li+后進行優化,得到冠醚-Li+復合物的穩定結構。
冠醚-Li+復合物中的O—Li+配位鍵電子密度AIM[29]拓撲分析使用了Multiwfn程序[30]。
冠醚與Li+之間的相互作用能(ΔEint)基于式(1)計算:
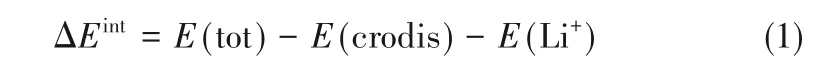
式中,E(tot)代表冠醚-Li+絡合物的電子能量,E(crodis)和E(Li+)分別代表形變后的冠醚和Li+的電子能量。在計算E(crodis)和E(Li+)時,冠醚和Li+的位置保持與復合物中一致。ΔEint計算在B3LYPD3(BJ)/Def2QZVP級別進行,其中,對所有原子采用了更大的Def2QZVP[27-28]基函數以提高計算精度,并通過Counterpoise方法[31]考慮了基組重疊誤差(BSSE)。
冠醚的幾何形變能(ΔEgeom)基于式(2)計算:
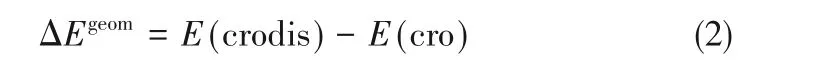
式中,E(cro)代表自由的冠醚的電子能量。
采用了自然布居分析(NPA)[32]和Mulliken[33-35]兩種電荷劃分方式計算原子電荷。
利用Multiwfn程序獲得了電子密度差和形變后的兩種冠醚分子范德華表面(isodensity=0.001 a.u.)靜電勢分布的空間格點,并利用VMD軟件[36]繪制了電子密度差圖(isovalue=0.006 a.u.)和靜電勢著色圖。電子密度差(Δρ)基于式(3)計算:
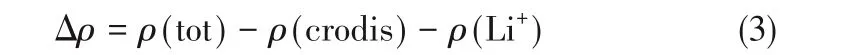
式中,ρ(tot)、ρ(crodis)和ρ(Li+)分別代表絡合物、形變后的冠醚和Li+的電子密度。
基于Morokuma-Ziegler方案[37-39]的能量分解分析使用了ADF程序[40],分別在BLYP-D3(BJ)/QZ4P[23-24,41]和B3LYP-D3(BJ)/TZP兩種理論級別下進行。該級別得到的ΔEint均與B3LYP-D3(BJ)/Def2QZVP級別結果符合很好。在該方案中,將ΔEint分解為式(4):
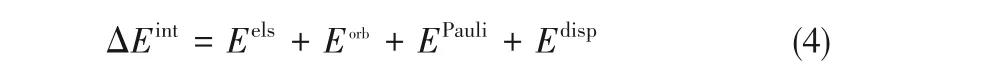
式(4)右邊四項分別代表靜電相互作用、軌道相互作用、Pauli排斥作用和色散作用。其中,Eels描述冠醚-Li+絡合物中,冠醚與Li+的電子密度互不影響的情況下,相互之間的靜電作用;Eorb通常用來描述軌道極化產生的共價相互作用,亦適用于描述本文中的O—Li+配位共價作用;EPauli用于描述冠醚與Li+之間,由于占據態軌道間Pauli排斥作用導致的絡合物整體能量升高;Edisp用來描述冠醚與Li+之間的色散相互作用。
溶劑條件下的絡合反應Gibbs自由能變使用Gaussian軟件基于式(5)~式(7)計算:

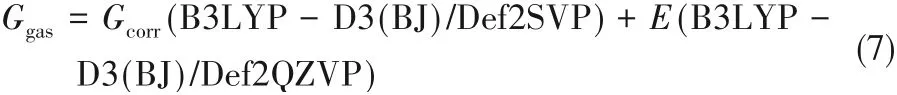
式中,Gsol、Esol和Ggas、Egas分別表示相應物質在溶劑條件下的Gibbs自由能、電子能量和在氣相條件下的Gibbs自由能、電子能量,Gcorr表示Gibbs自由能的熱力學矯正量,通過氣相條件下的頻率計算得到。Esol的計算在M052X/6-31G(d)[42-44]級別結合SMD溶劑[45]模型進行。在Gibbs自由能變計算中,所有物理量的單位統一為kcal/mol(1 kcal=4.18 kJ)。
2 結果與討論
圖2展示了5種自由冠醚以及它們絡合Li+之后的優化幾何結構。5個體系的詳細幾何參數展示在表1中,其中C—C、C—O、Si—O、Si—C和Si—Si分別表示相應化學鍵鍵長的平均值,C—Li+和Si—Li+為復合物中C和Si與Li+之間距離的平均值,On—Li+(n=1,2,3,4)分別為絡合物中相應O與Li+形成的配位鍵鍵長,O的編號與圖2相對應。表1中最后一行還給出了O—Li+配位鍵長的平均值。結合圖2和表1可知,5種冠醚中的C—C、C—O鍵長都十分接近。冠醚與Li+結合使C—C及Si—Si鍵長略有增加,而C—O和Si—O鍵長增量更為明顯。復合物中的C—Li+、Si—Li+較大(分別在2.75?和3.1?左右),而O—Li+較小(在1.9?左右)。分子結構圖中可以看出O與Li+有明顯的相互靠近趨勢。以上一系列結構特點表明Li+主要和O發生相互作用。優化所得的5種冠醚-Li+復合物均為Li+四配位模式,其中L、LSi、LSi-O和LSi-M與Li+為O—Li+四配位結構已經在晶體結構解析中得到了證實[16-17,46],而LSi/C-Li+晶體目前還未見相關報道。與Li+相互作用使冠醚的環結構發生了微弱的伸展,但是Si—C鍵長有所縮短,表明由于冠醚的形變及與Li+的相互作用,O與Si—O反鍵軌道的超共軛作用被削弱。
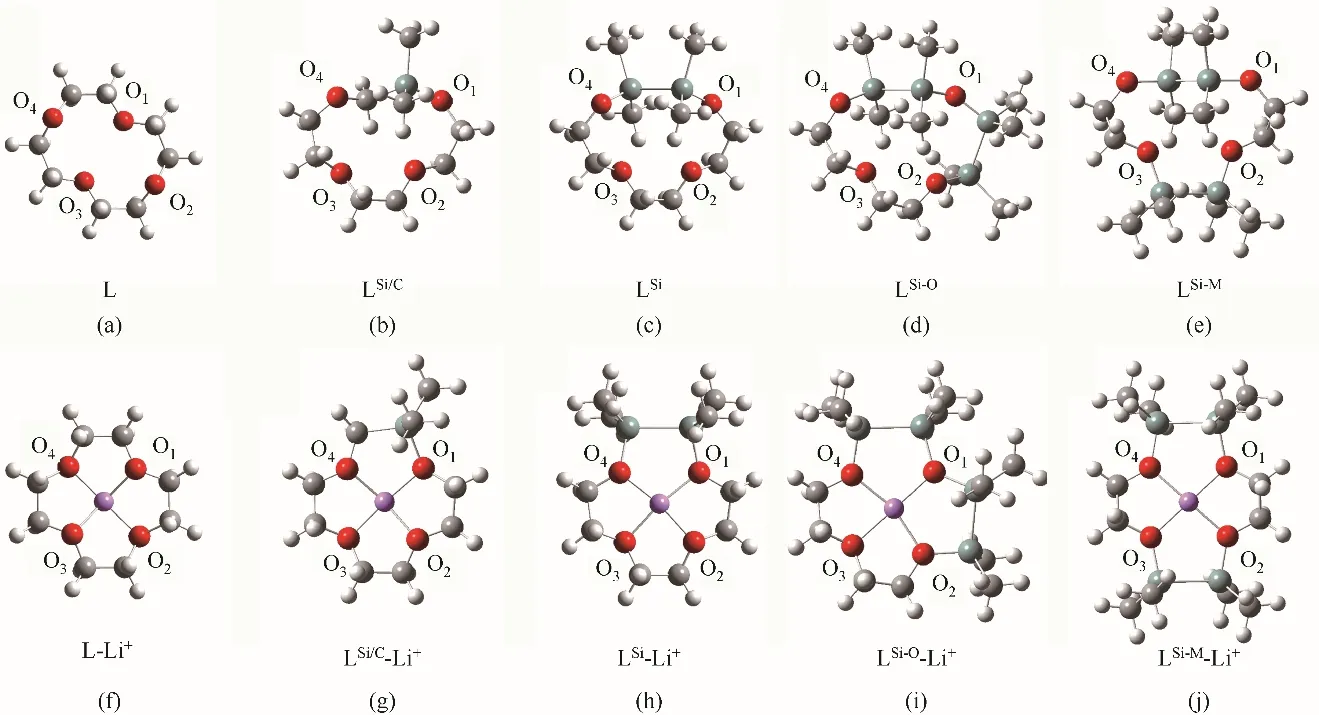
圖2 DFT優化的穩定分子幾何構象(灰色、紅色、白色、青色和紫色球體分別代表C、O、H、Si和Li原子)Fig.2 DFToptimized geometric structures
通過表1可以看出,在L-Li+中,4個O—Li+配位鍵鍵長幾乎相等(1.87~1.89?),這是由于L中4個O的化學環境本質上是等價的。在LSi/C-Li+中,O1與一個Si相鄰,而O1—Li+的數值較小(約1.72?),與之處于對位的O3—Li+數值較大(約1.91?)。經驗上講,配位鍵長通常與相互作用強度負相關。因此,可以認為LSi/C-Li+中的O1受Si的影響,與Li+之間產生了比另外三個O更強的相互作用。LSi-Li+中,由于O1和O4靠近Si,O2和O3遠離Si,這兩類O與Li的配位鍵長有差異。其中,靠近Si的O1/O4—Li+的鍵長(約1.89?)比遠離Si的O2/O3—Li+的鍵長(約1.94 ?)更短。Si使相鄰的O—Li+配位鍵長更短這一現象與LSi/C-Li+中觀察到的一致。但是,與L相比,—SiMe2—SiMe2—基團的引入使得LSi-Li+中的O—Li+平均配位鍵長增大了約0.04?(1.88?vs1.92?)。繼續增加摻雜Si數量到4,由于冠醚的尺寸增加,O—Li+平均配位鍵長被進一步拉長,并且LSi-O-Li+與LSi-M-Li+兩種情況下O—Li+配位鍵長存在差異(1.99 ?vs1.96?)。在LSi-O-Li+中,與1個Si相鄰的O4—Li+(約1.96?)配位鍵長最短,其次是與1個Si相鄰的O2—Li+(約1.98?)和與2個Si相鄰的O1—Li+(1.99 ?)。在LSi-M-Li+中,4個O—Li+配位鍵長相等(約1.96?),這是由于4個O的化學環境本質上是等價的。
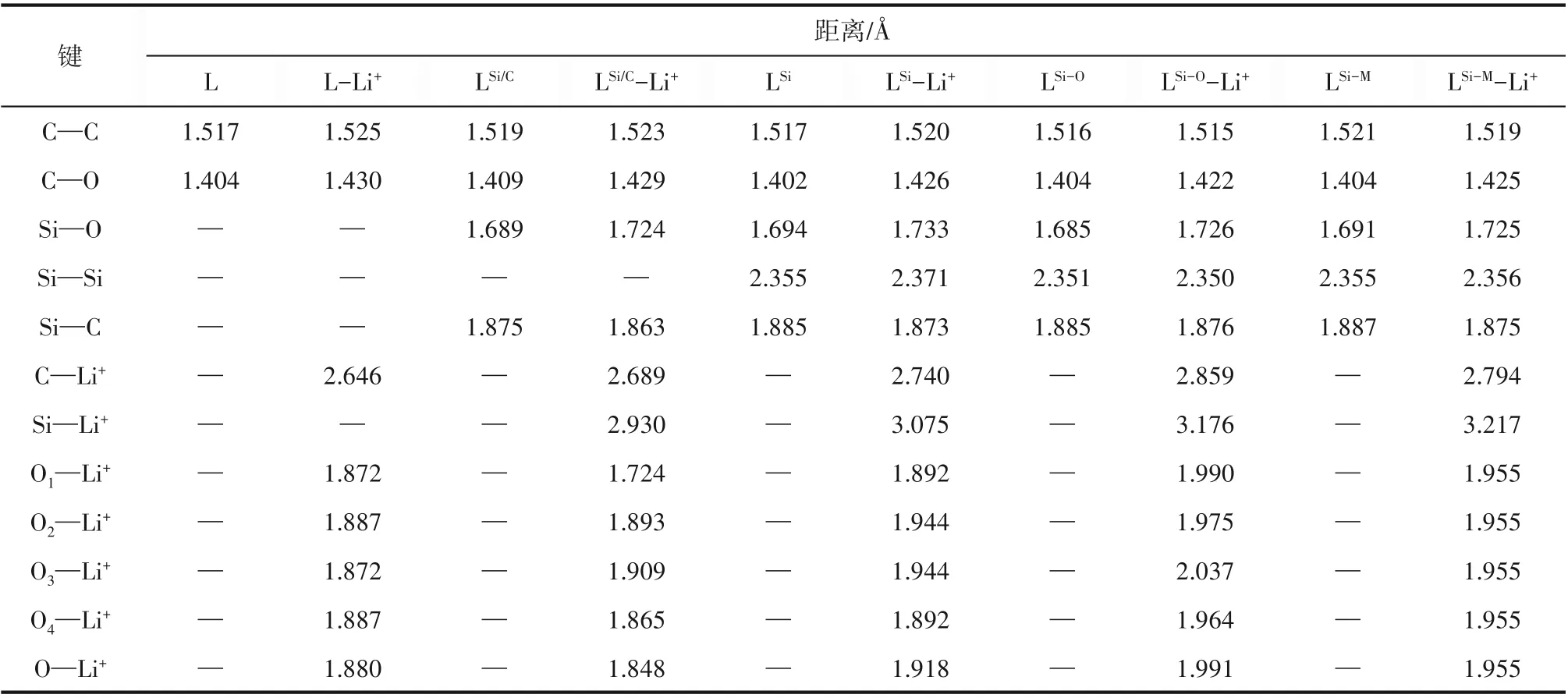
表1 自由冠醚及相應的冠醚-Li+絡合物的結構參數Table 1 Geometrical parameters of the free crown ethers and the crown-Li+complexes
O—Li+配位鍵的電子密度AIM拓撲分析用于揭示成鍵本質。在每一個O—Li+配位鍵鍵徑上都找到了鍵臨界點(BCP),并分析了該位置上的電子密度(ρ)、電子密度的拉普拉斯函數(?2ρ)、電子能量密度(H)和電子勢能密度絕對值(|V|)與動能密度(G)的比值(|V|/G),相關數據展示在表2中。對于共價作用,BCP處通常?2ρ<0、H<0、|V|/G>2,且ρ>0.14 a.u.;對于閉殼層作用(非共價作用),BCP處通常?2ρ>0、H>0、|V|/G<1,且ρ<0.14 a.u.[47-49]。通過表2中的數據可以看出,所有O-Li+的BCP處電子密度都很小(約0.03 a.u.),且?2ρ>0、H>0、|V|/G<1,具有明顯的非共價作用特征。因此,5種冠醚與Li+形成的O—Li+配位鍵本質上是閉殼層相互作用。

表2 O—Li+配位鍵BCP處電子密度拓撲性質Table 2 Topological electron density properties at critical points of the O—Li+
為了定量地比較5種冠醚對Li+的結合能力,在3種高精度級別下計算了冠醚-Li+的相互作用能,如表3所示。結果顯示,隨著摻雜Si原子數量的增加,冠醚與Li+的相互作用逐步增強,且摻雜Si的相對位置也會對冠醚與Li+的相互作用產生影響。LSi/C-Li+、LSi-Li+、LSi-O-Li+、LSi-M-Li+相互作用能分別強于L-Li+約8、14、20和24 kcal/mol,表明以—SiMe2—SiMe2—單元取代—CH2—CH2—單元確實可以顯著提升Si雜冠醚對Li+的絡合能力,與H?nisch等[16]的研究結論一致,但與LSi、LSi-O、LSi-M三種Si雜冠醚中更長的O—Li+配位鍵長相悖。另外,LSi/C-Li+相互作用比LSi-Li+更強,說明在不改變冠醚大環原子數的情況下摻雜單個Si也能夠提升冠醚對Li+的絡合能力。考慮到冠醚的形變需要消耗能量,計算了L、LSi/C、LSi、LSi-O和LSi-M的形變能,分別為11.26、16.72、19.28、23.73和23.57 kcal/mol。隨著Si摻雜數量越來越多而越來越大的形變能一定程度上削弱了Si雜冠醚對Li+的絡合能力,但即便將形變能扣除,LSi/C、LSi、LSi-O和LSi-M較L仍有約2、6、7和12 kcal/mol的優勢。
為了深入理解5種冠醚與Li+相互作用差異的來源,在BLYP-D3(BJ)/QZ4P和B3LYP-D3(BJ)/TZP級別計算了總相互作用能并進行能量分解分析,如表3所示。對表3縱向比較可知,5種冠醚與Li+的相互作用中靜電相互作用對總的相互作用能貢獻最大,軌道相互作用次之,Pauli互斥作用(正值)對相互作用起反貢獻,色散作用占比較低。通過對表3橫向比較,LSi/C-Li+、LSi-Li+、LSi-O-Li+和LSi-M-Li+的靜電相互作用分別比L-Li+高4~5、6~7、4~6和10~11 kcal/mol,表明Si摻雜顯著提升了冠醚-Li+的靜電相互作用,且提升效果受到摻雜Si的相對位置的影響;LSi/C-Li+、LSi-Li+和LSi-M-Li+的軌道相互作用與LLi+相差不到1 kcal/mol,LSi-O-Li+的軌道相互作用比L-Li+弱2~4 kcal/mol,表明幾何結構上的O—Li+鍵的拉長會略微削弱配位共價作用;對于不利于絡合的Pauli互斥作用,LSi/C-Li+、LSi-Li+、LSi-O-Li+和LSi-M-Li+分別比L-Li+小1~2、6~7、15~16和12~13 kcal/mol,這主要由于總體上摻雜Si使復合物中Li+距離Si雜冠醚更遠,也是Si雜冠醚對Li+絡合能力更強的另一個主導因素;相比于L-Li+,Si雜冠醚-Li+中的色散相互作用也體現出一定優勢(1~3 kcal/mol),這是由于Si元素的引入導致的。
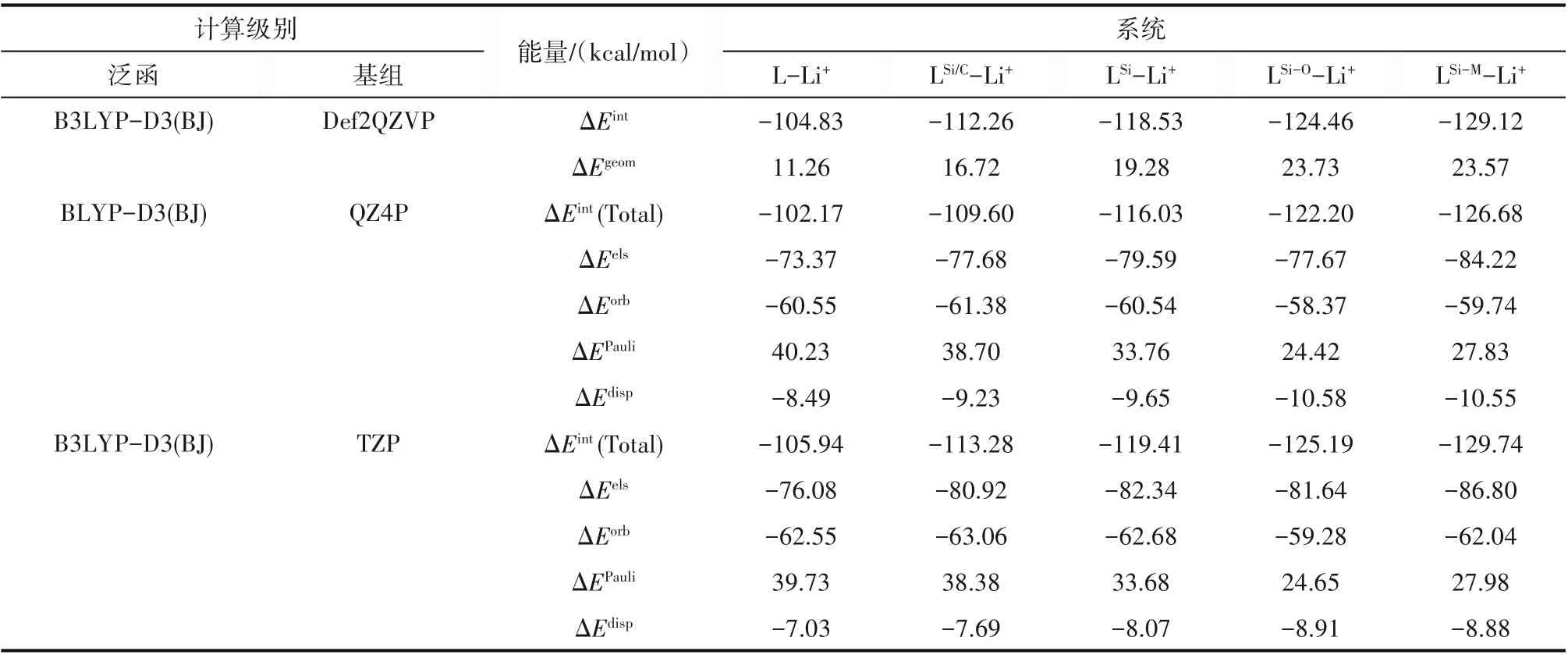
表3 冠醚-Li+相互作用能及能量分解Table 3 Crown-Li+interaction energies and energy decompositions
綜上分析,Si雜原子對冠醚與Li+之間靜電相互作用的增強作用和對Pauli互斥的削弱作用聯合主導了5種冠醚與Li+的相互作用差異,另外色散相互作用也有一定的貢獻。其中,靜電作用顯著增強的機理仍有待進一步研究。
進一步,分析了5個冠醚-Li+體系中冠醚與Li+之間的電子密度差,以及5種形變后的冠醚分子的范德華表面靜電勢分布,如圖3所示。這兩種基于體系基態電子密度波函數的方法可以真實并直觀地反映軌道相互作用和靜電相互作用。
從圖3的電子密度差圖中可見,在5種冠醚-Li+體系中,均有顯著的由O向Li+的電子密度極化(綠色),符合O—Li+配位鍵的化學本質。對比發現,除了LSi-O-Li+體系中O3—Li+鍵徑上綠色等值面包裹的區域明顯較小,其他4個體系中O的電子密度向Li+極化的程度十分相近。這也表明了,L-Li+、LSi/C-Li+、LSi-Li+和LSi-M-Li+4個體系中的軌道相互作用強度相近,而LSi-O-Li+體系中的軌道相互作用被較長的O—Li+配位鍵削弱,與前文能量分解的數據相符。
從圖3的電子密度范德華表面的靜電勢分布圖中可見,5種形變后的冠醚分子范德華表面的負靜電勢(紅色區域)主要分布于冠醚環的4個O原子附近。形變后的L、LSi/C、LSi、LSi-O和LSi-M的表面靜電勢極小值分別是-69.98、-76.50、-81.27、-89.25和-91.36 kcal/mol,由此可見Si的引入顯著誘導了O產生更負的靜電勢。進一步將兩種形變的冠醚O原子對范德華表面負靜電勢的貢獻劃分在以5 kcal/mol的連續區間內,并對每個區間求面積。可見,5種冠醚的所有O原子對負靜電勢的總貢獻(總面積)是相當的,但是隨著摻雜Si的數量增加,靜電勢的分布越來越偏向于靜電勢絕對值更大的區域,因此摻雜Si使靜電相互作用顯著增強。另外,結合能量分解的數據可知,實際上摻雜4個Si的LSi-O與Li+的靜電相互作用強度與摻雜1個Si的LSi/C相近,且弱于同樣摻雜4個Si的LSi-M約6 kcal/mol。這一現象在靜電勢極小值及O對靜電勢的貢獻情況中未得到充分體現。通過結構參數中的Si—Li+一項可知,與LSi-M相比,LSi-O中的Li+與Si的距離更小。因此可以推測,一方面,LSi-O中只有3個O和Si相鄰,使得Si對負靜電勢的增強作用沒能得到充分凸顯;另一方面,LSi-O-Li+中Si和Li+的距離較小,使得Si—Li+靜電互斥作用在更大程度上抵消了O對Li+的靜電吸引作用。
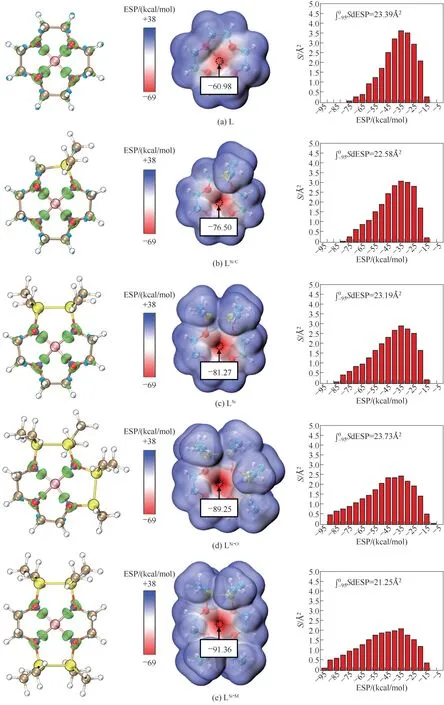
圖3 電子密度差圖和電子密度范德華表面(isodensity=0.001 a.u.)的靜電勢分布圖。左:電子密度差圖(isovalue=0.006 a.u.),綠色和藍色區域分別代表電子密度的積聚和衰減;中:形變后的冠醚分子的電子密度范德華表面的靜電勢分布圖;右:O對分子的電子密度范德華表面的靜電勢分布的貢獻Fig.3 Electron density difference diagramsand electrostatic potential distributions on the electron density van der Waals surface(isodensity=0.001 a.u.)
進而,通過表4的電荷布居分析可知,Si雜原子的引入使Si雜冠醚-Li+中的4個O原子所帶的負電荷總量明顯大于L-Li+,而這些負電荷增量更主要來自于靠近Si原子的O。由此可見,Si雜冠醚中的O從電負性低的Si得到了更多的電子,因此在Si雜冠醚的環中產生了更低的靜電勢,從而能夠與Li+產生更強的靜電吸引。而LSi-M-Li+中4個O所帶的負電荷總量大于LSi-OLi+,說明摻雜Si的數量相同時,冠醚中與Si相鄰的O數量越多,從Si得到的負電荷總量就越大。
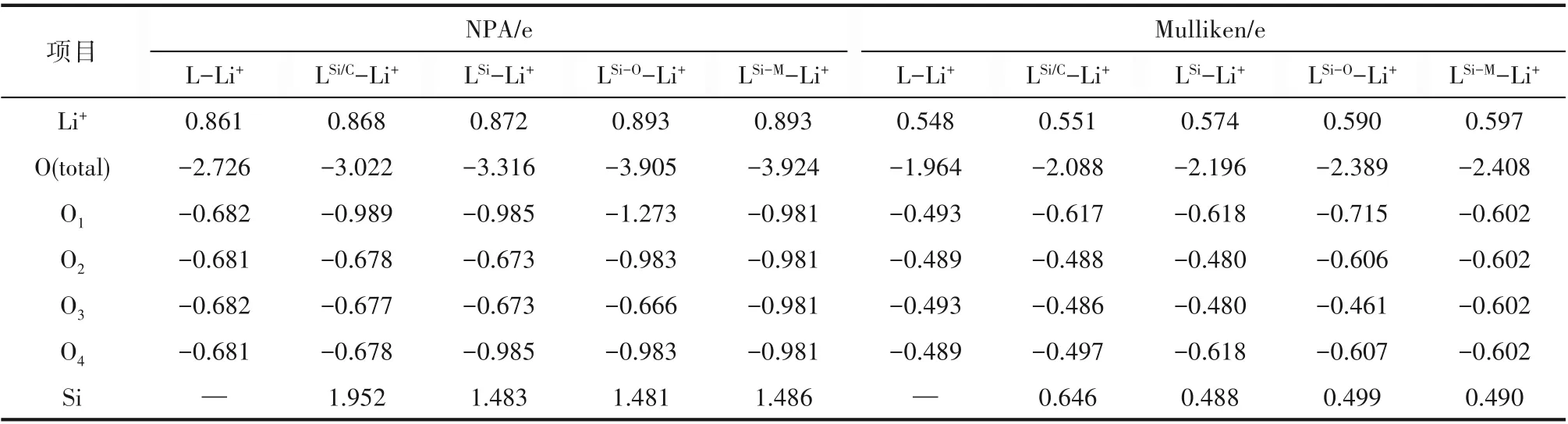
表4 冠醚-鋰離子絡合物中的電荷布居Table 4 Population analysis of crown-Li+complexes
Cameron等[15]的研究表明,由于Si轉移大量電子給了O,帶正電荷的Si與Li+之間具有庫侖排斥,因此Si雜冠醚對金屬離子的靜電相互作用可能比原始有機冠醚更弱。但在本文所考慮的以—SiMe2—SiMe2—單 元 和—CH2—SiMe2—替 換—CH2—CH2—的Si雜12-冠-4與Li+的絡合物中,Si能夠自發地遠離金屬Li+,從而有效避免了Si-Li+庫侖排斥,使得O原子的靜電優勢得以充分地凸顯。
冠醚通常用于離子的分離、檢測和反應催化,因此,冠醚-離子絡合反應一般在溶劑環境下進行。計算了5種冠醚在氣相條件下和二氯甲烷(DCM)溶劑條件下與Li+發生絡合反應的Gibbs自由能變,結果展示在圖4中。在氣相條件下LSi/C、LSi、LSi-O和LSi-M與Li+的絡合反應自由能變較L分別有約6、9、11和18 kcal/mol的優勢。而在DCM中,LSi/C、LSi、LSi-O和LSi-M與Li+的絡合反應自由能變較L分別有約3、5、6和11 kcal/mol的優勢。該趨勢與氣相條件下相互作用能的趨勢一致,說明摻雜Si原子所加強的冠醚-Li+相互作用將對溶液中冠醚-Li+絡合反應產生貢獻。此外,從Gibbs自由能變來看,DCM溶劑條件下的冠醚-Li+絡合反應比氣相條件下弱70~78 kcal/mol。這主要是由于Li+會與溶劑分子形成團簇,而冠醚絡合Li+導致一部分溶劑分子被脫除,這個過程會消耗能量。
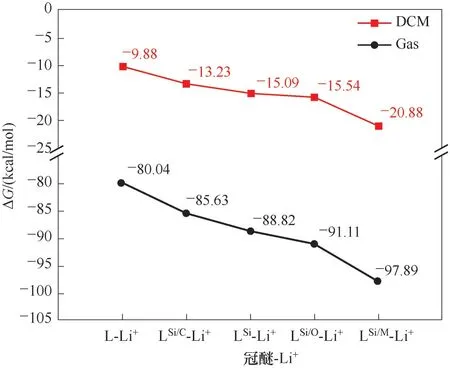
圖4 冠醚-Li+絡合反應的Gibbs自由能變Fig.4 Gibbs free energy changes of crown ether-Li+complexation reactions
3 結 論
本文基于量子化學密度泛函理論計算探究了12-冠-4(L)、1,1-2甲基-1-硅雜12-冠-4(LSi/C)、1,1,2,2-4甲基-1,2-2硅雜12-冠-4(LSi)、1,1,2,2,4,4,5,5-8甲基-1,2,4,5-4硅雜12-冠-4(LSi-O)和1,1,2,2,7,7,8,8-8甲基-1,2,7,8-4硅雜12-冠-4(LSi-M)與Li+的相互作用。結果表明,雖然LSi-Li+、LSi-O-Li+和LSi-M-Li+3種Si雜冠醚-Li+復合物比L-Li+具有更長的O—Li+配位鍵長,但是本工作中所研究的4種Si雜冠醚均具有對Li+更強的結合能力。根據結構參數、電子密度AIM拓撲分析、能量分解、電子密度差、靜電勢和電荷布居等一系列分析得出,O—Li+配位鍵具有閉殼層相互作用的特征。在LSi-Li+、LSi-O-Li+和LSi-M-Li+中,更長的O—Li+配位鍵長實際上僅略微削弱了配位鍵。Si原子由于電負性更弱而向O轉移了更多電子使O帶有更多負電荷,同時在絡合物中Si能夠自發地遠離Li+,從而有效地避免了Si—Li+庫侖排斥,降低了Si雜冠醚與Li+之間的Pauli排斥,充分凸顯了Si雜冠醚中O的靜電作用的優勢。當摻雜了2組—SiMe2—SiMe2—時,LSi-M與Li+的相互作用比LSi-O更強,說明使更多O與Si相鄰能令O得到更多負電荷。此外,Si的引入也使得冠醚與Li+之間的色散作用有一定程度增強。二氯甲烷中冠醚與Li+絡合反應的Gibbs自由能變說明,由摻雜Si帶來的相互作用能增強Si冠醚在二氯甲烷溶劑條件下絡合Li+的能力。因此,為了得到與金屬離子相互作用更強的Si雜冠醚,在分子合成中需要遵循以下3個規則:一是摻雜Si時不改變冠醚大環原子數;二是保證冠醚空穴和目標離子的尺寸相匹配;三是使盡量多的O與Si相鄰。
本工作揭示了Si雜原子提升冠醚對Li+絡合能力的根本機理,理清了一直以來對Si摻雜方式調控冠醚絡合金屬能力的困惑,對未來設計與合成具有高效萃取能力及其他特定功能的雜原子冠醚提供了必要的理論基礎。
符號說明
Edisp,Eels,Eorb,
Epauli——分別為色散相互作用能、靜電相互作用能、軌道相互作用能和Pauli互斥作用能,kcal/molE(tot),E(crodis),
E(Li+)——分別為冠醚-Li+絡合物的電子能量、形變后的冠醚和Li+的電子能量,kcal/mol
Egas,Esol——分別為相應結構在氣相條件下和溶劑環境中的電子能量,kcal/mol
ΔEdis,ΔEint——分別為形變能、相互作用能,kcal/mol
ESP——電子密度范德華表面的靜電勢,kcal/mol
G——電子的動能密度,a.u.
Gcorr——Gibbs自由能的熱力學校正值,kcal/mol
Ggas,Gsol——分別為相應結構在氣相條件下和溶劑環境中的Gibbs自由能,kcal/mol
ΔG——絡合反應Gibbs自由能變,kcal/mol
H——電子的能量密度,a.u.
S——電子密度范德華表面的面積,?2
V——電子的勢能密度,a.u.
ρ,Δρ——分別為電子密度、電子密度差,a.u.
ρ(tot),ρ(cro),
ρ(Li+)——分別為絡合物電子密度、形變后冠醚的電子密度和鋰離子的電子密度,a.u.
?2ρ——電子密度的拉普拉斯函數,a.u.
