牛病毒性腹瀉病毒非結構蛋白p7 的克隆和原核表達純化
2021-06-17 02:59:26付強郭妍婷楊莉史慧君
中國畜禽種業 2021年5期
付強 郭妍婷 楊莉 史慧君
(新疆農業大學動物醫學學院 830052)
牛病毒性腹瀉病毒(Bovine Viral Diarrhea Virus,BVDV)屬于黃病毒科(Flaviviridae)、瘟病毒屬(Pestivirus),主要感染牛、羊、駱駝等家畜和野生動物[1],是牛病毒性腹瀉-黏膜病(Bovine Viral Diarrhea-Mucosal Disease,BVD-MD)的主要病原之一[2]。該病毒感染成年動物后常呈現隱性感染,一般不表現明顯的臨床癥狀,而幼畜感染后常造成腹瀉、高燒、白細胞減少、黏膜糜爛甚至脫落等癥狀,感染孕畜后造成流產、產死胎和畸形胎、繁殖障礙等[3,4]。尤其是非致細胞病變型(Non-cytopathogenic Effect,NCP)BVDV 感染妊娠早期母畜,造成持續性感染(Persistent Infection,PI)犢牛的出現,PI 犢牛一旦再次感染CP 型BVDV 后表現出嚴重的腹瀉黏膜病,最終因脫水和代謝性酸中毒等致死率高達90%,而且PI 犢牛始終向外排毒,成為污染整個牛群的重要傳染源[5,6]。
BVDV 非結構p7 蛋白在病毒復制過程中起到重要的作用。有研究報道,非結構蛋白p7 翻譯后折疊并加工成六聚體形式,轉運至細胞膜上,組成離子孔道,所以p7 蛋白屬于離子孔道蛋白家族,主要參與介導一些離子(Na+、Ca2+、K+、H+等)運輸,而且對病毒進入細胞及子代病毒在細胞中成熟產生影響[7-9]。本研究開展BVDV 非結構蛋白p7 的基因克隆及原核表達純化,以期獲得p7 蛋白,為后續構建p7 的多克隆和單克隆抗體提供重要的材料和平臺。
1 材料和方法
1.1 細菌、載體和cDNA
BL21(DE3)PLySs 大腸桿菌感受態細胞購自北京索萊寶科技有限公司;pCold-MBP 載體購自武漢淼靈生物科技有限公司;BVDV 毒株NADL 的cDNA 保存于新疆農業大學動物醫學學院動物病毒實驗室。
1.2 主要試劑和儀器
考馬斯亮藍快速染色液、透析袋和5×蛋白上樣緩沖液購自北京索萊寶科技有限公司;Kpn I 和BamH I 購自NEB 公司;Taq PCR Mastermix、T4 Ligase、瓊脂糖凝膠DNA 回收試劑盒和普通質粒提取試劑盒購自天根生化科技(北京)有限公司;Ni-TED 瓊脂糖樹脂、NaH2PO4、NaCl、咪唑imidazole、瓊脂糖和異丙基-b-D-硫代半乳糖苷IPTG 購自生工生物工程(上海)股份有限公司;低溫高速臺式離心機Micro 21R 購自美國Thermo 公司;C1000 PCR 儀購自美國BioRad 公司;DYCP-31DN型核酸瓊脂糖水平電泳儀購自北京六一生物科技有限公司。
1.3 p7 基因克隆及測序分析
根據GenBank 數據庫中BVDV NADL(NC_001461.1)基因組序列中p7 基因,設計擴增p7 基因引物為p7-F:5’CGGggtaccATGTCCCAGTATGGGGCAGGTG 3’;p7-R:5’ CG CggatccAGCCT TTGCCATCCCTCCAAC 3’,其中ggtacc 和ggatcc分別為Kpn I 和BamH I 酶切位點。以BVDV NADL cDNA 為模版,以p7-F 和p7-R 為引物,PCR 擴增p7 基因,PCR 反應體系見表1,PCR 反應條件為95℃5min;95℃30s、60℃30s、72℃30s,35 個循環;72℃10min;4℃∞。使用瓊脂糖凝膠電泳檢測PCR 結果,并使用瓊脂糖凝膠DNA 回收試劑盒回收p7 基因,將p7 基因送至蘇州金唯智生物科技有限公司進行測序分析。
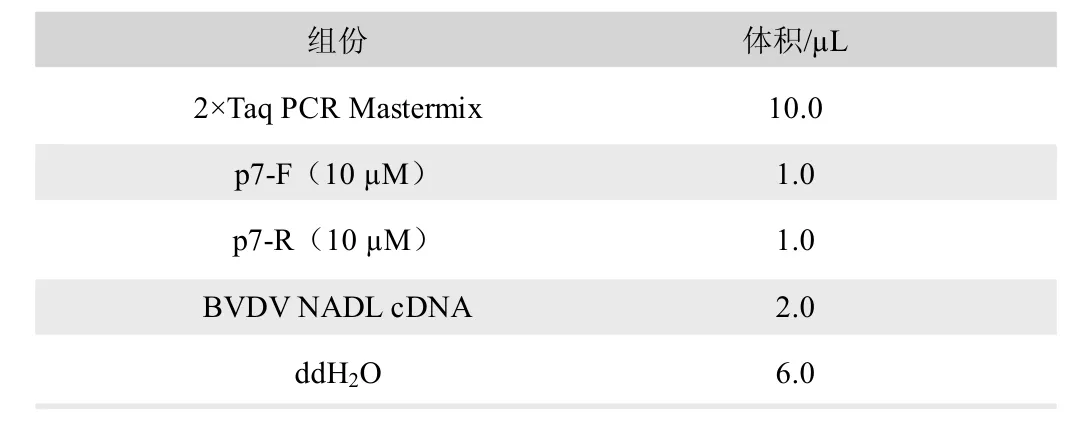
表1 PCR 反應體系
1.4 pCold-MBP-p7 原核表達載體構建
使用Kpn I 和BamH I 對p7 基因和pCold-MBP 質粒進行酶切,酶切體系見表2,酶切反應條件為37℃4 h。酶切反應完成后使用瓊脂糖凝膠電泳進行檢測,并使用瓊脂糖凝膠DNA回收試劑盒將酶切后的p7 基因和pCold-MBP 載體回收,使用T4 DNA Ligase 將p7 基因和pCold-MBP 載體連接,連接反應條件見表3,連接反應條件為16℃過夜;將連接產物熱激轉化至BL21(DE3)PLySs 大腸桿菌感受態細胞,涂布到含有氨芐西林的瓊脂平板中,37℃培養過夜,次日使用PCR 鑒定單克隆菌落,并將陽性克隆送至蘇州金唯智生物科技有限公司進行測序鑒定。
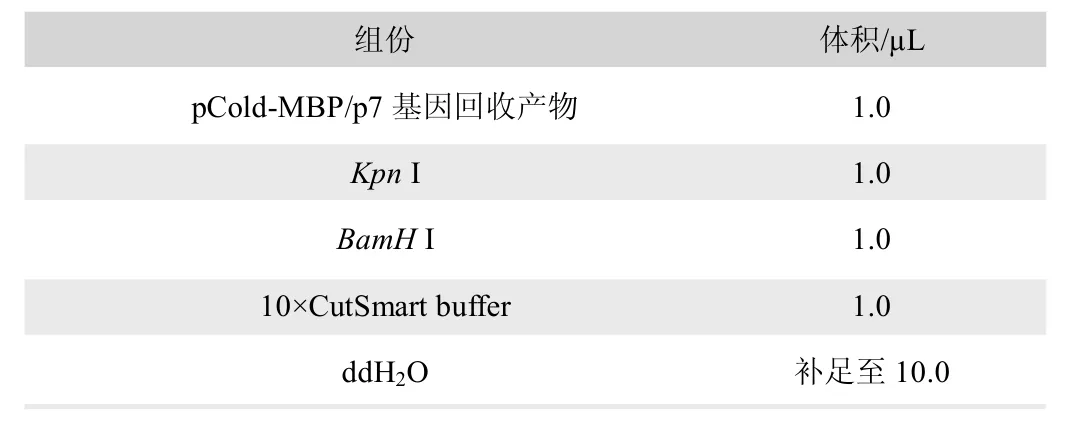
表2 酶切反應體系
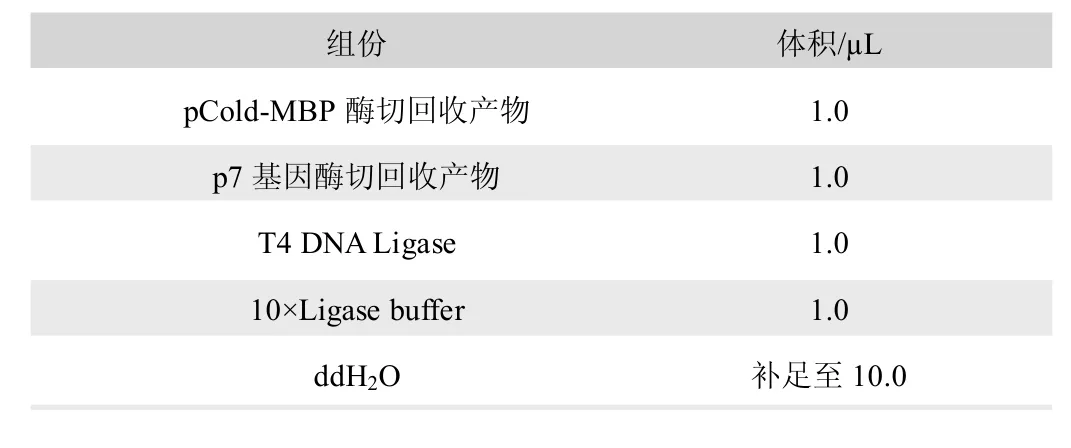
表3 連接反應體系
1.5 IPTG 誘導條件優化
將pCold-MBP-p7 單克隆菌落接種至LB 肉湯培養基中擴繁,擴繁完成后以體積比1:100 比例接種至新的LB 肉湯培養基中,置于37℃200rpm 振蕩培養,待OD600 nm 為0.4~0.6時,分別加入終濃度0、0.6、0.8、1.0、1.2 mM IPTG,16℃條件下200rpm 振蕩過夜誘導表達;次日分別取1ml 培養物,8000rpm 離心5min 后棄掉上清,加入100μL 1×蛋白上樣緩沖液,使用SDS-PAGE 電泳分離蛋白質,并使用考馬斯亮藍染色套裝(染色液+脫色液)進行染色和脫色,具體染色和脫色步驟見試劑盒說明書。
1.6 pCold-MBP-p7 原核表達載體誘導表達和純化
pCold-MBP-p7 單克隆菌落擴繁步驟同上,擴繁后以體積比1:100 比例接種菌液至新的LB 肉湯培養基中,置于37℃200rpm 振蕩培養,待OD600nm為0.4~0.6 時,加入終濃度為1.2 mM IPTG,置于16℃低溫過夜誘導表達,次日離心收集菌體沉淀,加入10ml 裂解液(50 mM NaH2PO4,500mM NaCl,20mM imidazole,調節pH 至8.0)重懸沉淀,使用細胞超聲破碎儀(15s ON,15s OFF)進行超聲破碎至液體清亮。8,000rpm 離心10min 去除細胞碎片,填加到Ni-TED 瓊脂糖樹脂中,4℃孵育4h 后使用3 倍柱體積的洗滌緩沖液(50mM NaH2PO4,500mM NaCl,40mM imidazole,調節pH 至8.0)進行洗滌,加入洗脫緩沖液(50mM NaH2PO4,300mM NaCl,250mM imidazole,調節pH 至8.0)進行4 次洗脫,收集洗脫溶液,進行SDS-PAGE電泳并使用考馬斯亮藍溶液染色進行觀察。
1.7 蛋白透析及檢測
將透析袋裁剪成適當長度,置于2% NaHCO3和1mM EDTA 溶液中煮沸10min;使用蒸餾水清晰透析袋后,使用1mM EDTA 溶液煮沸透析袋10min,待冷卻后灌裝洗脫蛋白溶液,置于100 倍體積的磷酸鹽緩沖液(20mM,pH 7.0)放置于4℃下滲透過夜,次日轉移剩余蛋白溶液至15ml 離心管,取少量蛋白溶液進行SDS-PAGE 電泳,使用考馬斯亮藍溶液染色進行觀察,其余置于-80℃為后續使用。
2 結果
2.1 p7 基因擴增及pCold-MBP-p7 原核表達載體構建
以BVDV NADL cDNA 為模版,使用PCR 擴增p7 基因。如圖1 所示,PCR 產物大小約為225bp,與p7 基因大小一致;將p7 基因克隆至pCold-MBP 載體后,獲得pCold-MBP-p7 原核表達載體,質粒圖譜見圖2。
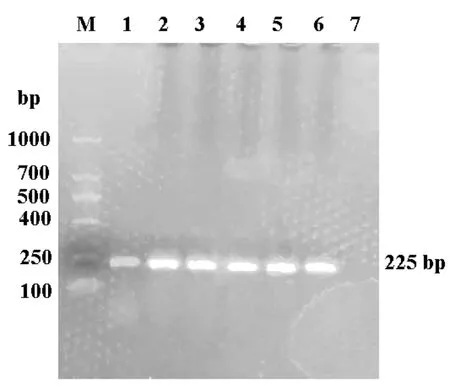
圖1 PCR 擴增p7 基因結果

圖2 pCold-MBP-p7 原核表達載體圖譜
2.2 IPTG 誘導條件優化
加入不同濃度的IPTG 后低溫過夜誘導,次日收集菌體并使用SDS-PAGE 進行檢測。結果如圖3 所示,使用1.2mM IPTG 誘導后能蛋白表達量最高,所以后續試驗使用1.2mM IPTG 作為誘導條件。
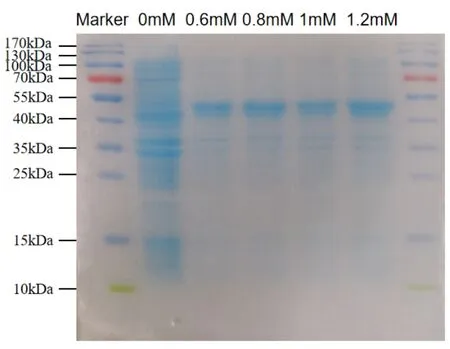
圖3 IPTG 誘導濃度優化
2.3 pCold-MBP-p7 原核表達和蛋白純化
使用IPTG 誘導pCold-MBP-p7 表達過夜,收集菌體后破碎,使用Ni-TED 瓊脂糖樹脂進行純化。結果如圖4 所示,MBP-p7 融合蛋白成功表達,約49kDa;經過4 次洗脫液洗脫后大部蛋白已從瓊脂糖樹脂洗脫下來,得到雜帶較少的MBPp7 融合蛋白。
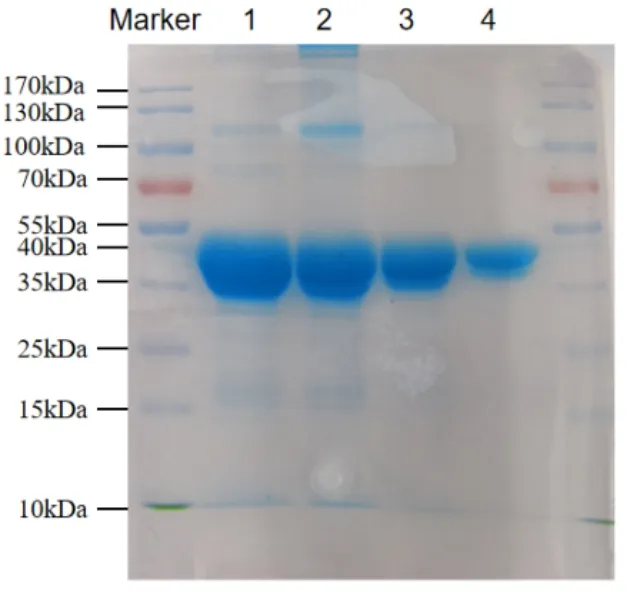
圖4 MBP-p7 蛋白純化
2.4 MBP-p7 蛋白透析
使用透析袋對MBP-p7 蛋白進行透析,完成后使用SDS-PAGE進行電泳,使用考馬斯亮藍染色進行鑒定。結果如圖5 所示,透析后MBP-p7 蛋白條帶較為單一,表明MBP-p7 蛋白純度高。

圖5 MBP-p7 蛋白透析后鑒定
3 討論
黃病毒科非結構蛋白p7 經翻譯后加工聚集并轉運至細胞膜上形成六聚體離子孔道形式,參與到離子跨膜運輸、病毒侵入和釋放等過程,對病毒復制起到重要作用[10]。而目前關于p7 蛋白如何進行聚集,在什么時間p7 蛋白開始聚集尚不清楚,本課題組正在開展BVDV p7 形成離子孔道的機制研究,而目前尚未見BVDV p7 蛋白相關的抗體銷售,開展本試驗表達純化p7 蛋白,擬為后續p7 蛋白的多克隆抗體及單克隆抗體研發提供重要的材料。
本試驗使用pCold-MBP 原核表達載體進行誘導表達純化,其中pCold 屬于冷休克基因CSPA 啟動子,在較低溫度下即可誘導蛋白表達,進而降低蛋白誘導過程中因溫度過高造成的蛋白聚集、變性、形成難溶物等情況發生[11];MBP 蛋白標簽是一種麥芽糖結合蛋白標簽,分子量約42.5 kDa,一般情況下將MBP 蛋白融合在目的蛋白的N 端,有利于減少目的蛋白的降解,提高蛋白的可溶性,維持蛋白結構穩定性,減少純化難度[12]。本試驗使用pCold 實現低溫高效誘導表達,使用MBP 蛋白標簽實現維持蛋白穩定性的作用。
