氯喹通過阻斷腫瘤壞死因子受體相關因子3自噬性降解抑制類風濕關節炎滑膜細胞活化
2021-05-28 05:59:10呂昌偉和晶張乾郗海濤強毅張靜濤
實用醫學雜志 2021年8期
呂昌偉 和晶 張乾 郗海濤 強毅 張靜濤
西北大學附屬醫院/西安市第三醫院骨科(西安710018)
類風濕性關節炎是一種慢性炎癥性自身免疫性疾病,最終可導致骨關節損傷甚至殘疾[1]。類風濕性關節炎病理改變以慢性滑膜炎和骨關節結構損害為特征,其發病機制尚未明確,目前并無完全有效的根治方法[2]。近來的研究顯示,纖維樣滑膜細胞(fibroblast?like synoviocyte,FLS)的異常活化在類風濕性關節炎的發展中發揮了關鍵作用,其可通過產生炎性因子引起滑膜炎癥和病理損傷[3]。活化的FLS表現出過度增殖,并產生多種炎癥因子、趨化因子和基質金屬蛋白酶(matrix metalloproteinase,MMP),如TNF?α、IL?1β、IL?6、CXCL10、MMP?2和MMP?9,造成關節的慢性炎癥和持續性破壞[4]。腫瘤壞死因子受體相關因子3(TNF receptor associated factor 3,TRAF3)是屬于腫瘤壞死因子受體作用因子超家族成員,是細胞內一種重要的負性調控蛋白,包括抑制CREB、NF?κB和STAT3信號[5]。NF?κB信號的異常激活可引起持續高強度炎癥,導致IL?6、MMP?2、MMP?9、CXCL10等多種炎性因子水平升高,誘發多種自身免疫性疾病[6],并在FLS的激活中發揮了重要作用[4]。
目前,氯喹用于類風濕性關節炎治療取得了較好的療效,但其作用機制尚不明確。本研究探討氯喹對FLS異常激活的抑制作用,并通過探討氯喹對TRAF3蛋白水平的影響,以及對TRAF3自噬性降解的影響,以期闡明氯喹抑制FLS活化的分子機制,為氯喹治療類風濕性關節炎的臨床應用提供依據。
1 材料與方法
1.1 細胞培養與處理類風濕關節炎滑膜細胞(MH7A)于重慶巴而思生物科技有限公司購買,采用RPMI?1640培養基(Gibco,美國)補充10%胎牛血清(AusGeneX,澳大利亞)和1%青鏈霉素(碧云天,中國)進行培養。采用TNF?α預處理建立類風濕關節炎滑膜細胞活化模型,即除對照組外,所有MH7A細胞接受5 ng/mL的重組人THF?α(BBI,中國)預刺激24 h。根據實驗,分別采用20 μmol/L氯喹(MCE,美國)、10 μmol/L雷帕霉素(Rapa,MCE)和10 μmol/L蛋白酶體抑制劑MG?132(MCE)處理細胞24 h。siRNA干擾TRAF3表達細胞,進行siRNA轉染后,進行上述藥物處理。
1.2 si?RNA轉染使用不含青霉素/鏈霉素的RPMI?1640培養基接種細胞至6孔板,培養12 h。將TRAF3 siRNA(Santa Cruz,美國)溶于optiMEM培養基(Gibco),將相應體積lipofectamin 2000(In?vitrogen,美國)溶于optiMEM培養基中,將配制好的siRNA溶液和lipofectamin 2000溶液等體積混合,室溫穩定20 min,加入4倍體積新鮮optiMEM培養基混勻。吸出細胞舊培養基,每孔加入1 mL配制好的siRNA轉染液,細胞培養箱中孵育5 h,換為正常培養基,繼續培養24 h后用于后續實驗。
1.3 細胞增殖檢測細胞處理結束后,采用CCK?8試劑(碧云天)檢測細胞增殖。吸出舊培養接,使用新培養基配制CCK?8溶液,加入各處理孔,37℃培養箱放置1 h,使用酶標儀(TECAN,瑞士)在450 nm測定吸光度。
1.4 Real?time PCR細胞處理結束后,RNAiso Plus(TaKaRa,中國)按照使用說明書提取總RNA,采用PrimeScriptRT Master Mix試劑盒(TaKaRa)按照說明書逆轉錄為cDNA。采用TB Green Premix Ex Taq II試劑盒(TaKaRa)按照使用說明,進行real?time PCR檢測。引物序列如下:GAPDH:forward 5′?AGGTCGGTGTGAACGGATT?3′,reverse 5′?AAT CTCCACTTTGCCACTGC?3′;TRAF3:forward 5′?AC?ATCCGCCTAGCCGACATGG?3′,reverse 5′?CTGCT?TCCGCCGCTTGTAGTC?3′。
1.5 ELISA檢測細胞處理結束后,收集細胞培養上清,采用ELISA試劑盒檢測細胞培養上清中促炎因子含量。IL?6和CXCL10 ELISA檢測試劑盒訂購于博士德生物,按照產品說明書進行操作。
1.6 Western blot細胞處理結束后,RIPA裂解液(碧云天,中國)提取蛋白,BCA蛋白測定試劑盒(碧云天)測定蛋白濃度,加入4×Laemmli上樣緩沖液(Bio?Rad,美國),97.5℃變性10 min。SDS?PAGE凝膠電泳,轉模后5%脫脂奶粉室溫封閉,洗膜后分別孵TRAF3一抗(1∶1 000,CST,美國)、LC3一抗(1∶1 000,CST)、MMP?2一抗(1∶1 000,Protein?tech)、MMP?9一抗(1∶1 000,Proteintech),β?actin一抗(1∶2 000,CST),4℃過夜。洗膜后羊抗鼠和羊抗兔二抗(1∶1 000,碧云天)室溫孵育1 h,化學發光,ChemiDocTMXRS+凝膠成像系統(Bio?Rad)中曝光檢測。
1.7 統計學方法結果數據采用()表示,采用GraphPad Prism 8.0軟件進行統計分析。兩組數據之間的比較采用t檢驗,多組數據之間的比較采用單因素方差分析并跟以Fisher′s post hoc檢驗進行兩兩比較。P<0.05為差異有統計學意義。
2 結果
2.1 氯喹抑制類風濕關節炎滑膜細胞活化相比對照組,TNF?α刺激導致了MH7A細胞IL?6和CX?CL10分泌顯著增加(圖1B、C),并且引起了MH7A細胞侵襲蛋白MMP?2和MMP?9表達增加(圖1D),表明TNF?α預刺激有效引起了MH7A細胞活化。與TNF組相比,氯喹處理顯著抑制了MH7A細胞增殖(圖1A),降低了TNF?α引起的IL?6和CXCL10分泌(圖1B、C),以及MMP?2和MMP?9表達(圖1D)。這些結果表明,氯喹可顯著抑制類風濕關節炎滑膜細胞活化。
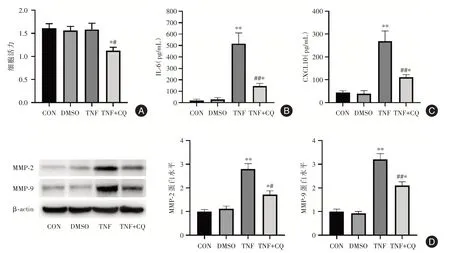
圖1 氯喹抑制MH7A纖維樣滑膜細胞活化Fig.1 Chloroquine inhibits the activation of MH7A fibroblast?like synoviocytes
2.2 氯喹引起MH7A細胞TRAF3蛋白含量升高TNF?α刺激和氯喹處理均未引起MH7A細胞TRAF3 mRNA表達的變化(圖2A),表明氯喹并不引起TRAF3基因表達水平的改變。然而,Western blot結果顯示。氯喹處理可導致MH7A細胞內TRAF3蛋白含量顯著升高(圖2B)。這些結果提示,氯喹所致的TRAF3蛋白水平升高,并不是源于其基因表達的上調,而可能源于氯喹抑制了TRAF3的降解。
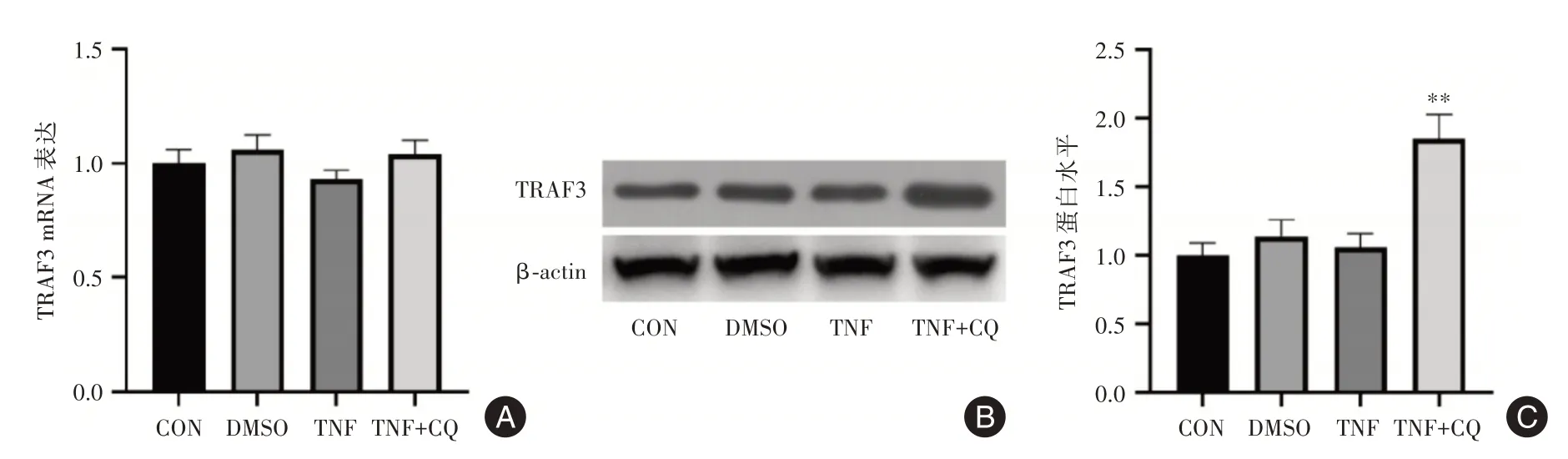
圖2 氯喹引起MH7A滑膜細胞TRAF3蛋白表達升高Fig.2 Chloroquine increased the expression of TRAF3 protein in MH7A synoviocytes
2.3 TRAF3介導了氯喹對MH7A細胞活化的抑制通過TRAF3 siRNA下調MH7A細胞TRAF3蛋白表達,探討TRAF3在氯喹抑制MH7A細胞活化中的作用。結果顯示,si?TRAF3顯著降低了MH7A細胞內TRAF3蛋白水平(圖3A)。由結果可見,si?TRAF3可顯著拮抗氯喹對MH7A細胞活化的抑制作用,阻斷了氯喹對IL?6和CXCL10的分泌抑制(圖3B、C),并解除了氯喹對MMP?2和MMP?9蛋白表達的抑制(圖3D)。
2.4 氯喹抑制了TRAF3蛋白的自噬性降解進一步實驗可見,氯喹可引起MH7A細胞LC3Ⅱ/Ⅰ蛋白比值降低(圖4A),表明氯喹抑制了MH7A細胞自噬。自噬誘導劑雷帕霉素(RA)則顯著增加了細胞自噬水平(圖4A)。并且,雷帕霉素拮抗了氯喹的效應,導致MH7A細胞TRAF3蛋白水平明顯降低(圖4B)。此外,氯喹所致的TRAF3蛋白含量升高未被蛋白酶體抑制劑MG132進一步增強(圖4B)。這些結果表明,氯喹可能主要通過抑制細胞自噬,阻斷TRAF3的自噬性降解,導致了MH7A細胞TRAF3蛋白含量升高。
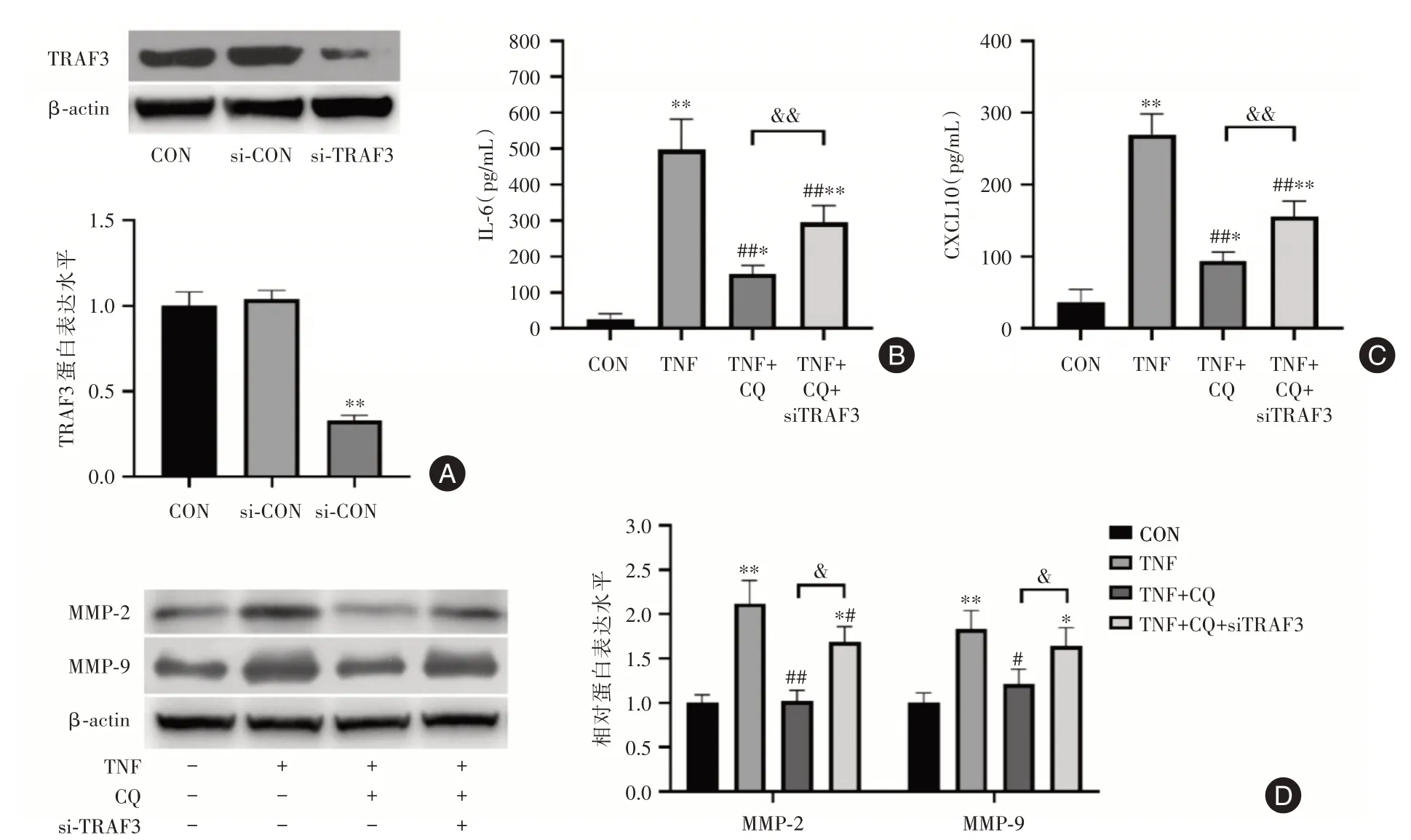
圖3 TRAF3介導了氯喹對MH7A細胞的活化抑制Fig.3 TRAF3 mediated the inhibition of the activation of MH7A cells by chloroquine
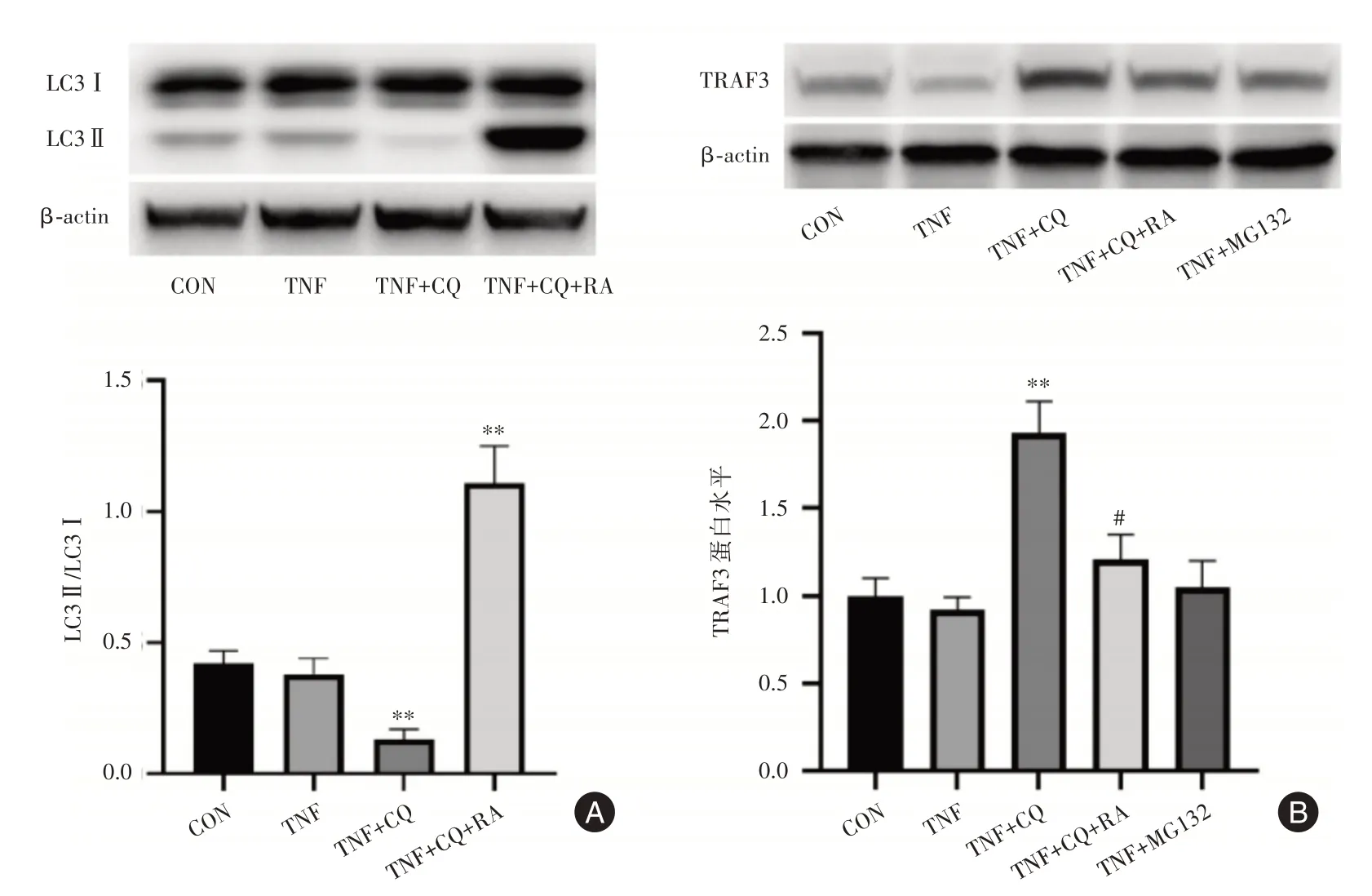
圖4 氯喹抑制MH7A細胞自噬和TRAF3蛋白自噬性降解Fig.4 Chloroquine inhibits autophagy of MH7A cells and autophagic degradation of TRAF3 protein
2.5 誘導自噬可拮抗氯喹對MH7A細胞的活化抑制采用自噬誘導劑雷帕霉素和氯喹共處理MH7A細胞。可見,與氯喹處理組相比,雷帕霉素可對抗氯喹作用,顯著促進MH7A細胞IL?6和CX?CL10分泌增加(圖5A,B),并引起MH7A細胞MMP?2和MMP?9蛋白水平升高(圖5C)。這些結果進一步證實了氯喹通過阻止TRAF3自噬性降解抑制MH7A細胞活化。
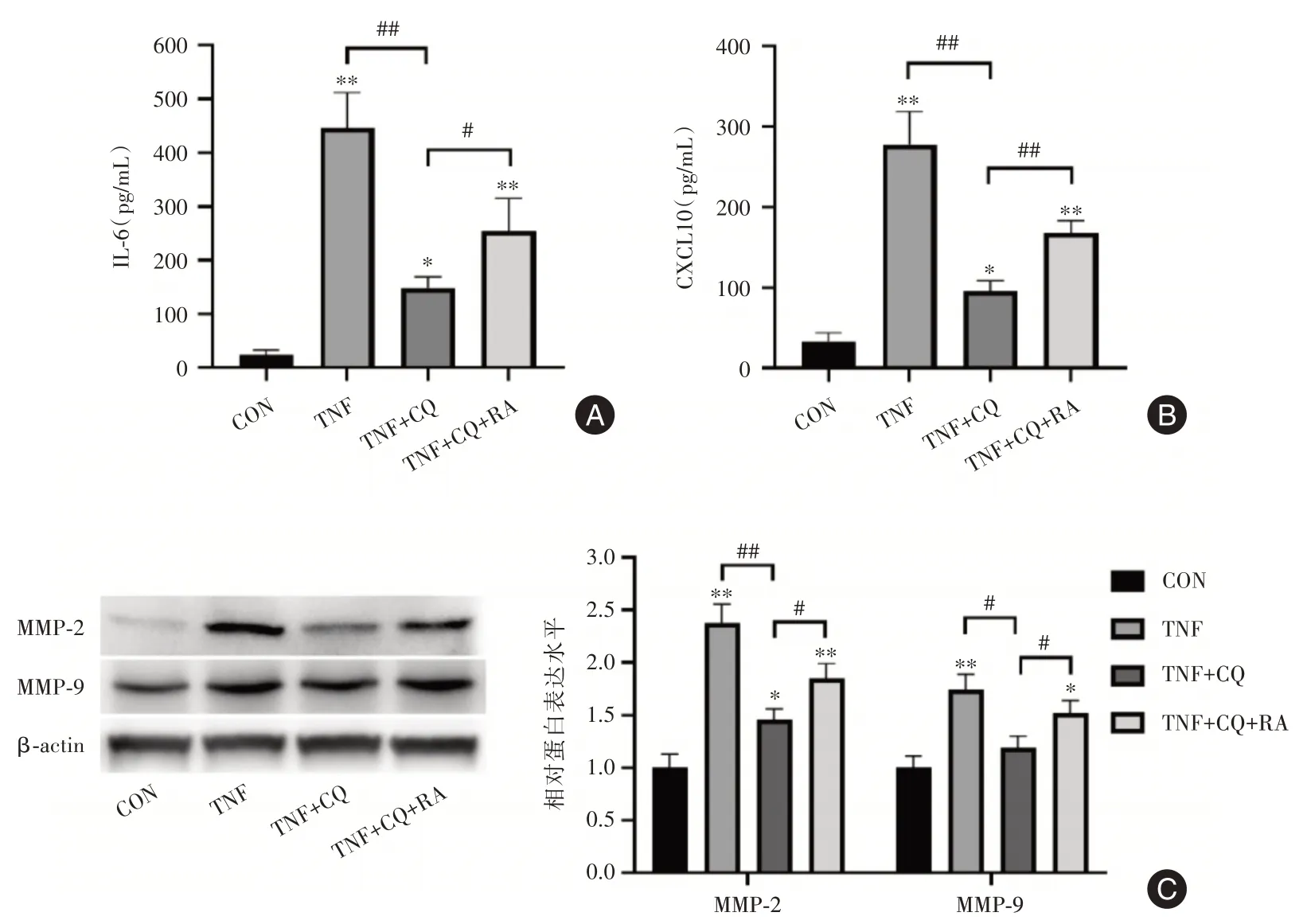
圖5 誘導自噬可拮抗氯喹對MH7A細胞的活化抑制Fig.5 Induction of autophagy antagonized the inhibition of MH7A cell activation by chloroquine
3 討論
類風濕性關節炎的病理機制至今尚未明確,使得其缺乏確切有效的根治方法。近來,纖維樣滑膜細胞(FLS)的異常活化在類風濕性關節炎的發病機理中獲得了重視。FLS在類風濕性關節炎的多種病理進程中均發揮了關鍵作用,如滑膜炎癥,血管翳生長,以及最終的軟骨和骨破壞[7]。FLS在炎性滑膜的異常增生是類風濕性關節炎的典型特征[7]。這種細胞的增多源于FLS的大量增殖和凋亡減少[8]。另外,FLS被激活后可分泌基質金屬蛋白酶,使細胞表現出侵襲特性,并導致關節組織破壞[7,9]。在類風濕性關節炎的滑膜炎癥中,FLS激活同樣發揮了重要作用。活化的FLS可分泌多種促炎因子,如IL?6、IL?1β和CXCL10等[10-11],進而影響其他細胞和自身免疫反應性。本研究中,采用TNF?α預刺激建立FLS活化模型。結果可見,TNF?α預刺激可引起FLS細胞增殖,導致MMP?2和MMP?9蛋白表達升高,并引起IL?6和CXCL10分泌的增加,表明TNF?α刺激有效引起了FLS細胞的活化。
氯喹和羥氯喹已被應用于治療輕中度類風濕性關節炎,以及重度類風濕性關節炎治療時的聯合用藥[12]。雖然氯喹對免疫?炎癥反應的抑制作用已被發現和報道,包括抑制免疫細胞增殖和活性,抑制炎癥因子分泌,但對其治療類風濕性關節炎的藥理機制了解仍非常有限[13]。在本實驗研究發現,氯喹處理可有效抑制FLS細胞的增殖、炎癥因子分泌以及侵襲標志物MMP?2和MMP?9的表達,表明氯喹可有效抑制FLS的異常活化。這些結果表明,抑制FLS異常激活可能是氯喹治療類風濕性關節炎的重要機制。同時,氯喹是一種高效的親溶酶體抑制劑,可特異的抑制細胞溶酶體自噬途徑[14]。自噬從多個方面參與了類風濕性關節炎的病理進程,包括FLS的存活、侵襲和凋亡抵抗[15]。最新的研究顯示,抑制自噬可以提高類風濕性關節炎的治療效果[16]。因此,氯喹可能通過抑制溶酶體自噬而發揮上述FLS活化抑制作用。本研究證實,氯喹處理可顯著抑制FLS細胞自噬,表現為LC3Ⅱ/Ⅰ蛋白比值的降低。
TRAF3是一種重要的炎癥?免疫負性調控因子,其通過抑制多種信號通路而抑制免疫細胞活化,如NF?κB、IL?6R、CD40和CREB信號通路[5,17]。已有研究顯示,激活TRAF3信號可以抑制肝癌細胞的增殖和侵襲,并可減輕動物炎癥反應[18-19]。本研究發現,氯喹能夠增加MH7A細胞內TRAF3的蛋白含量,而干擾TRAF3表達阻斷了氯喹對MH7A細胞活化的抑制,表明氯喹通過TRAF3增加抑制FLS活化。進一步研究發現,并且這種增加不是由于基因表達的增加。因此,氯喹可能通過抑制溶酶體自噬降解而實現TRAF3的蛋白含量升高。這一猜測被自噬誘導劑和蛋白酶體抑制劑實驗證實。這些結果表明,氯喹通過阻斷TRAF3溶酶體自噬降解,抑制FLS細胞活化。
NF?κB信號激活和類風濕性關節炎的滑膜病變程度程正相關,抑制NF?κB表達和信號激活,可顯著抑制類風濕性關節炎的病情進展[20],而增加NF?κB表達可加速類風濕性關節炎的侵襲進展[21]。NF?κB是TRAF3的主要負性調控信號通路,因此TRAF3對纖維樣滑膜細胞活化的抑制可能藉由抑制NF?κB信號實現。
本研究從類風濕性關節炎纖維樣滑膜細胞異常活化角度,闡明了氯喹治療類風濕性關節炎的藥理機制。氯喹長時間大劑量的類風濕性關節炎治療應用可誘發不良反應,尤其是眼毒性[22]。因此,闡明氯喹治療類風濕性關節炎的藥理機制,有助于指導臨床聯合用藥,降低藥物劑量,有利于降低不良反應的發生。
