白介素在血管鈣化作用機制中的研究進展
2021-05-17 07:31:36張菲菲周子皓王芳
心血管病學進展 2021年4期
關鍵詞:研究
張菲菲 周子皓 王芳
(1.南京醫科大學第一臨床醫學院,江蘇 南京 210029; 2.南京醫科大學第一附屬醫院心血管內科,江蘇 南京 210029)
血管鈣化(vascular calcification,VC)是礦物質在血管中的病理性沉積,其與心血管疾病的死亡率高度相關,任何動脈壁中的鈣化都會使死亡和心血管事件的風險增加3~4倍,尤其是在患有糖尿病和慢性腎臟病的高危患者中[1]。造成VC的原因包括慢性炎癥、代謝和遺傳等,其中慢性炎癥扮演了極其重要的角色。眾多炎性細胞及炎性因子參與了這個過程,如巨噬細胞、單核細胞、白介素(interleukin,IL)-1β、IL-6和腫瘤壞死因子-α(tumor necrosis factor-α,TNF-α)等,但炎癥反應誘發及介導VC的分子機制尚未完全闡明。IL驅動VC的過程可通過介導內皮細胞或血管平滑肌細胞(vascular smooth muscle cell,VSMC)的成骨轉化,誘導VSMC衰老等發揮作用[2-3](表1)。因此,闡明IL家族成員在VC中的作用對臨床治療心血管疾病及其他VC相關疾病具有重大意義。
1 促炎性IL
1.1 IL-1β
IL-1β是促進VC的重要因子。2017年的一項回顧性臨床試驗表明,在動脈粥樣硬化患者和糖尿病患者鈣化的冠狀動脈中IL-1β水平升高,這些患者的冠狀動脈鈣負荷與血清IL-1β水平呈正相關,且心源性猝死的發生率也隨之增加[4],這一結果表明IL-1β可作為冠狀動脈疾病患者的生物標志物與鈣化風險和心血管終點的預測指標[5]。體外細胞實驗發現,IL-1β可與TNF-α共同誘導人主動脈內皮細胞發生間充質轉化,這是內皮細胞特征喪失和成纖維細胞表型獲得的過程,最終導致其成為具有成骨潛能的細胞,有助于VC[6]。巨噬細胞釋放IL-1β誘導VSMC發生成骨轉化后,VSMC會反過來刺激巨噬細胞產生更多的IL-1β,形成正反饋加速VSMC的成骨轉化,促進VC的發生[4]。近年的研究發現細胞衰老在VC中發揮重要作用,其中IL-1β參與了這一過程。作為終末期腎病患者VC的重要生物標志物,IL-1β在血管內側平滑肌中高表達,高水平的IL-1β通過激活核因子κB(nuclear factor-κB,NF-κB)/p53/p21通路誘導VSMC的衰老和成骨轉化,最終導致VC[7]。還有研究表明IL-1β的產生也可促進內皮細胞衰老[8]。目前已了解慢性低度炎癥在VC過程中起重要作用,同時這也是細胞衰老的重要特征,探究細胞衰老到VC過程中炎癥因子的作用,有助于對鈣化的進一步了解以及防治。
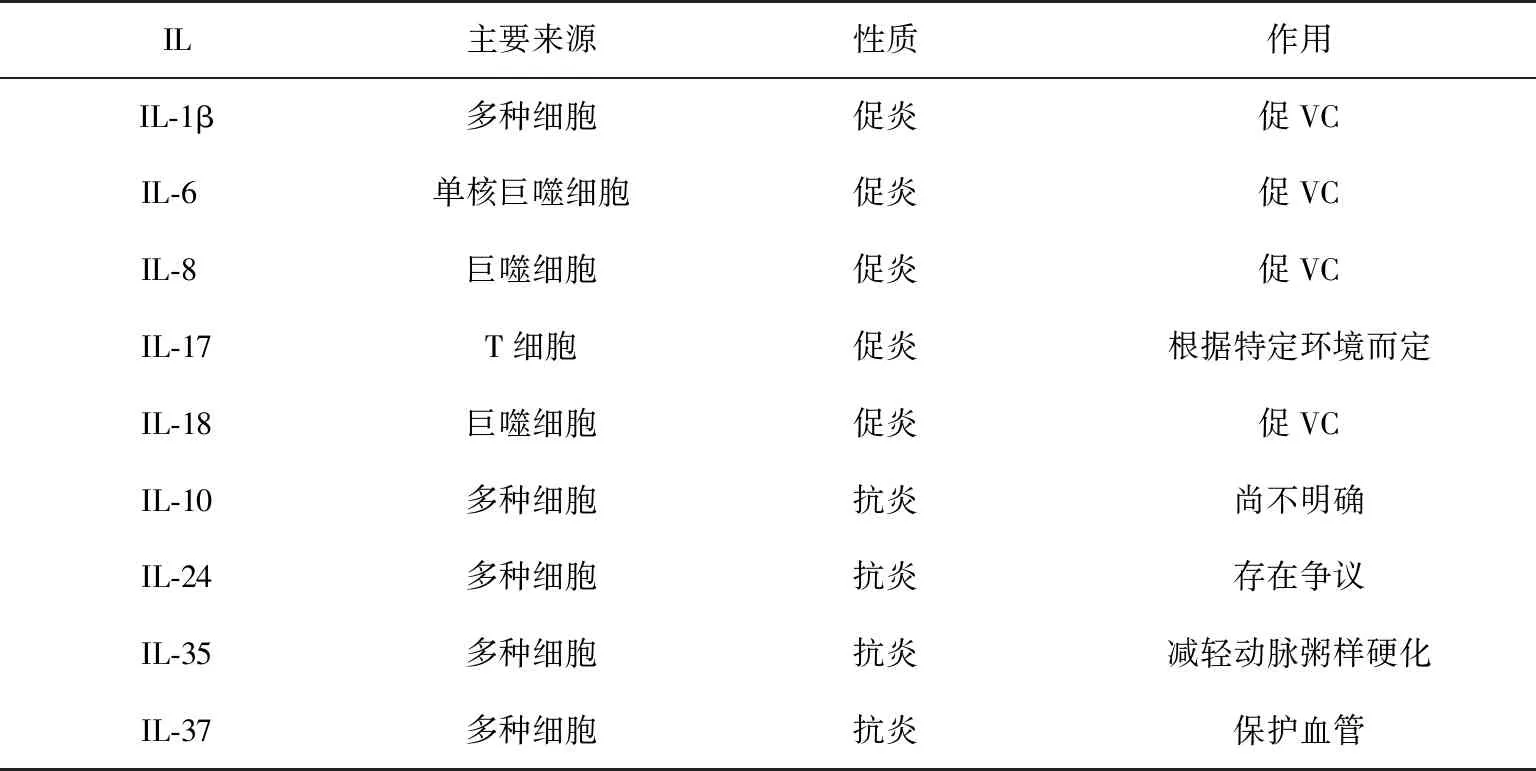
表1 IL與VC
1.2 IL-6
IL-6主要由單核巨噬細胞產生。IL-6高血清水平常伴隨心血管疾病高死亡率,可作為冠狀動脈疾病的死亡風險評估因子。VC的過程通常涉及內皮細胞或VSMC的成骨轉化,IL-6與骨保護/破壞的相關因子參與了這一過程。有研究表明,IL-6是NF-κB受體激活蛋白配體(receptor activator of NF-κB ligand,RANKL)、NF-κB受體激活蛋白和骨保護素(osteoprotegerin,OPG)的強誘導劑,且RANKL也可上調IL-6的表達[9]。2018年Lee等[10]研究發現Toll樣受體2在動脈粥樣硬化中促鈣化的作用,Toll樣受體2通過上調IL-6介導RANKL異常表達進而誘導VSMC鈣化和軟骨分化。Kurozumi等[11]第一次提出炎癥因子IL-6可強烈誘導人VSMC成骨轉化,在慢性炎癥相關的VC中具有一定作用。IL-6通過與可溶性IL-6受體(sIL-6R)結合激活p-STAT3,提高Runt相關轉錄因子2、堿性磷酸酶(alkaline phosphatase,ALP)以及骨橋蛋白的表達。除此之外,Xu等[12]發現在60例終末期腎病患者中,有56例患者血清IL-6和磷酸鹽水平升高,且其中有一半人存在心血管鈣化,IL-6和磷酸鹽過載可能促進終末期腎病患者的VC。除IL-1β外,IL-6也可促進VSMC衰老相關的鈣化。進一步的研究表明,IL-6可通過激活IL-6/sIL-6R/STAT3/P53/P21通路誘導終末期腎病患者血管平滑肌衰老相關的VC,且抗衰老劑減輕VC。細胞衰老有關的鈣化的具體機制值得科研人員去進一步探索。
1.3 IL-8
趨化因子是一種可誘導細胞發生定向遷移的小細胞因子或信號蛋白,可使白細胞轉移至損傷部位引發炎癥反應。IL-8作為趨化因子家族的成員之一,可作為冠心病患者和終末期腎病患者臨床預后的獨立預測因子[13]。近年來發現,IL-8是血管內皮細胞和VSMC功能的重要調節因子。在患有慢性腎臟疾病時,無機磷和吲哚硫酸酯等尿毒癥毒素會誘導內皮細胞IL-8的表達和分泌[14]。而IL-8則會誘導人VSMC的鈣化,且以一種濃度依賴性方式增強鈣化。但IL-8誘導鈣化的機制不是促進VSCM的成骨分化,而是通過阻遏鈣化抑制因子骨橋蛋白的表達驅動VC。
越來越多的趨化因子被證明在VC或動脈粥樣硬化的生理過程中扮演著重要的角色,但針對IL-8與VC的研究還比較少,除遏制骨橋蛋白,IL-8是否還通過其他途徑刺激VSMC以及內皮細胞來促進VC,需科研工作者繼續挖掘,為心血管疾病的治療提供新的思路及靶點。
1.4 IL-17
IL-17是由免疫細胞CD4+T細胞的亞型Th17細胞分泌的一種促炎癥細胞因子,參與人體眾多炎癥性和自身免疫性疾病。關于IL-17與血管疾病的研究目前大多集中在動脈粥樣硬化,而VC作為動脈粥樣硬化的一種病理現象,可由多種免疫細胞分泌的促炎癥因子的刺激引起。許多研究表明,IL-17作為一種促炎癥細胞因子在動脈粥樣硬化中扮演著重要的角色,其在動脈粥樣硬化中的多功能性仍存在爭議,因為IL-17對動脈粥樣硬化的促進或抑制取決于特定的細胞組織和免疫環境。
Orejudo等[15]研究表明IL-17A具有直接的血管作用,其作用機制似乎與誘導VSMC肥大和表型改變有關,但關于VC方面的研究目前較少。最新關于IL-17促VC的研究是基于川崎病患者的冠狀動脈鈣化[16]。體外研究表明,同時給予IL-17和γ干擾素(interferon-gamma,IFN-γ)誘導蛋白-10可觸發患者冠狀動脈平滑肌細胞鈣化,此過程通過誘導Runt相關轉錄因子2、ALP等成骨轉化因子的表達得以實現,IL-17和IFN-γ誘導蛋白-10單獨處理時,二者均不能誘導鈣化發生。它們是如何相互作用誘導鈣化的還有待研究,同時在正常人體內是否也存在類似的鈣化誘導機制也有待研究。近期一項關于IL-17和IFN-γ與原代成骨細胞的早期分化及鈣化的研究表明,用IL-17和IFN-γ直接對原代成骨細胞進行處理并未發現細胞數量增加,當使用IL-17和IFN-γ誘導培養基處理原代成骨細胞時,成骨細胞表現出高活性的ALP以及骨鈣素和OPG基因的高表達,但IL-17和IFN-γ卻對成骨細胞的鈣化起抑制作用[17]。IL-17在VC中扮演的角色目前尚不清楚,具體是抑制鈣化還是促進鈣化,以及何種環境下IL-17對鈣化作用發生改變,都需進一步研究。
1.5 IL-18
IL-18是一種促炎細胞因子,主要由巨噬細胞合成并釋放,VSMC也能釋放IL-18[18]。在VC方面,IL-18的研究相對較少。Zhang等[19]的研究發現,IL-18單獨作用時不會引起原代大鼠VSMC VC和血管壁細胞的成骨轉化,而在VC的誘導條件下,IL-18可加速鈣化和促進VSMC的成骨分化。M型瞬時受體電位是Mg2+和Ca2+的可滲透離子通道,與人類成骨細胞的增殖和遷移有關。IL-18通過EPK1/2信號通路激活M型瞬時受體電位,促進β-甘油磷酸酯(β-glycerophosphate,β-GP)誘導的VSMC的鈣化和成骨分化。Schelski等[18]也發現IL-18可增強磷酸鹽誘導的VSMC的骨/軟骨轉分化和鈣化,通過上調血清和糖皮質激素激酶誘導VSMC的成骨分化,糖皮質激素激酶是在鈣化過程中起關鍵作用的調控因子。不同的是,他們發現單獨用IL-18處理原代人主動脈VSMC可誘導成骨轉化,這與Zhang團隊的研究成果不一致,可能是由于種屬差異導致的不同結果,有待進一步探究。
2 抗炎性IL
2.1 IL-10
IL-10是一種由多源性細胞分泌的抗炎癥細胞因子,人體內的IL-10主要由輔助性T細胞和單核巨噬細胞分泌。作為抗炎細胞因子的IL-10在VC中的功能尚未明確。Huo等[20]發現動脈粥樣硬化患者外周血IL-10表達顯著低于正常人外周血中的水平。同時也有研究指出動脈粥樣硬化患者體內分泌IL-10的調節性B淋巴細胞的數量較正常人少,且與炎癥狀況呈負相關[21]。以上研究結果從側面說明患者外周血IL-10水平不足可能是導致鈣化的原因之一。也有研究對IL-10在VC中的功能提出異議,最直接的證據來自IL-10敲除的小鼠模型。Sage等[22]構建了一個具有IL-10 B淋巴細胞特異性缺乏癥低密度脂蛋白缺乏的小鼠模型,經過8周高脂飲食后,與對照組低密度脂蛋白缺乏的小鼠對比發現:兩組小鼠除動脈粥樣硬化斑塊的大小外,斑塊的組成及動脈硬化的程度均無差別,指出調節性B淋巴細胞分泌的IL-10在小鼠動脈粥樣硬化的發展中不是必需的。最新一項包含多種族930例患動脈粥樣硬化成人的調查報告也指出:在未患臨床心臟病的個體中,IL-10水平的高低似乎與是否發生心血管疾病無關,且是亞臨床冠狀動脈粥樣硬化的不良標志[23]。因此,IL-10作為一種抗炎因子在VC中的具體功能還存在爭議,需進一步明確。
2.2 IL-24
目前對IL-24在腫瘤方面的研究較多,而對其在心血管疾病方面的作用還知之甚少。2012年Lee等[24]研究稱:IL-24作為抗炎細胞因子可顯著抑制β-GP誘導的VSMC鈣化。它是通過抑制β-GP誘導的細胞凋亡和成骨標志物的表達來抑制VC的。IL-24阻斷β-GP誘導的Wnt/β-catenin通路的激活,該通路在VC的發病機制中起關鍵作用。在VC中,IL-24可通過Wnt/β-catenin途徑上調Runt相關轉錄因子2轉錄來刺激鈣化和成骨標志物的表達。提示IL-24抑制VSMC鈣化與Wnt/β-catenin途徑失活有關。表明IL-24是一種潛在的血管平滑肌鈣化治療劑。且2013年Lee等[25]又發現外源性給藥IL-24可減弱H2O2處理誘導的小鼠VSMC血管炎癥和高血壓相關基因的表達,提示IL-24基因可能是動脈粥樣硬化研究的重要候選基因。但2016年Kawada等[26]的研究結果與Lee完全相反。他們發現IL-24可刺激人主動脈平滑肌細胞發生鈣化,在此過程中使骨形態發生蛋白2的表達升高,且抗IL-24抗體可逆轉IL-24的作用,這一點表明IL-24在人VSMC鈣化中確實有重要作用[26]。但具體環境可能決定了IL-24對鈣化的不同作用,尤其是在體內環境中其與鈣化的關系需大量臨床數據去探究驗證。
2.3 IL-35
IL-35是IL-12細胞因子家族的新成員,自2007年發現以來很快成為研究熱點。IL-35可由單核細胞、T細胞、B淋巴細胞、Treg細胞及腫瘤細胞等多種類型的細胞表達,通過激活JAK-STAT信號通路參與免疫功能的調節,與轉化生長因子β和IL-10一樣被認為是最重要的三種免疫抑制因子之一,對IL-35在自身免疫性疾病和腫瘤疾病中的研究報道較多。目前尚缺乏IL-35與VC的直接研究數據,但有人發現在ApoE-/-小鼠模型中,IL-35可減輕動脈粥樣硬化的血管損傷,同時主動脈內膜厚度和斑塊都得到明顯改善。王慶航等[27]提出IL-35抑制動脈粥樣硬化的作用可能由于對IL-10、轉化生長因子β和IL-17水平的調控。Li等[28]發現IL-35通過抑制內皮細胞激活相關分子細胞間黏附分子-1的表達,來抑制未刺激的原代人單核細胞與溶血磷脂酰膽堿處理的人主動脈內皮細胞的黏附,還可通過抑制線粒體活性氧的產生抑制內皮細胞激活。目前,IL-35在動脈粥樣硬化中的保護功能已被大多數研究證實,但其在VC中的作用還缺乏直接的證據。
2.4 IL-37
IL-37是新近發現的抗炎細胞因子,共有a~e五種不同的亞型,IL-37b是目前研究的熱點。IL-37主要由單核巨噬細胞、樹突狀細胞、內皮細胞和VSMC等分泌,可抑制體內多種促炎細胞因子的表達及增強抗炎細胞因子的活性。眾多研究表明IL-37在VC中具有保護血管的作用。
2014年Chai等[29]第一次發現IL-37在小鼠鈣化模型中可減輕VC,IL-37可上調抗炎因子IL-10的表達,并下調促炎因子IL-18和TNF-α的表達,由此猜測IL-37可能在VC中參與炎癥的調節和抗炎的介導。且在給予IL-37處理的小鼠體內觀察到了OPG的升高,而給予抗OPG抗體以后,IL-37對血管的保護作用減小且伴隨ALP和骨形態發生蛋白2的表達增加,表明IL-37的血管保護作用部分依賴于OPG[30]。在一項200例志愿者參與的臨床研究中,觀察到嚴重冠狀動脈鈣化組的患者血漿IL-37水平顯著升高,且與鈣化程度呈正相關,在此過程中也觀察到患者血漿OPG水平的升高,且與IL-37水平呈正相關[30]。近期有研究表明,重組IL-37通過Notch1/NF-κB途徑抑制M1型巨噬細胞極化而發揮抗炎作用[31-32]。而IL-37在VC中發揮抗炎作用的具體機制尚不清楚。
3 小結與展望
綜上所述,慢性炎癥是VC的重要環節。在此過程中,各類免疫細胞被體內環境刺激活化并分泌細胞因子。這些細胞因子通過各種受體及信號通路介導內皮細胞和/或VSMC的成骨分化及隨之而來的鈣化。在成骨分化過程中,炎癥因子誘導的細胞衰老和內皮細胞及VSMC的間充質轉化非常關鍵,抑制細胞衰老和間充質轉化可能成為未來治療VC的新靶點。同時具有抗炎作用的細胞因子的研究目前還存在一定爭議,但其在動脈粥樣硬化中的研究也提供了新的思路。動脈粥樣硬化、慢性腎病和糖尿病等與VC都存在慢性炎癥的作用,所以抗炎是治療這些慢性疾病的關鍵。但由于炎癥因子的作用比較復雜,在研究過程中會因研究試劑、模型的選擇等的差異導致研究結果的不同,所以需科研工作者們更加深入廣泛地研究去闡明炎癥因子作用的確切途徑,為VC提供有效的防治措施,降低心血管病的死亡率。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
