新疆兩種土地利用方式下土壤病毒的群落組成與功能特征
2021-04-25 06:26:14于丹婷張麗梅賀紀正韓麗麗
生態學報 2021年7期
畢 麗,杜 帥,于丹婷,張麗梅,賀紀正,韓麗麗,*
1 中國科學院生態環境研究中心,城市與區域生態國家重點實驗室, 北京 100085
2 中國科學院大學, 北京 100049
3 福建師范大學地理科學學院, 福州 350007
病毒是地球上數量最多、種類最豐富的生物實體,也是生態系統中食物鏈和食物網的重要組成部分。環境病毒生態學早期主要在海洋生態系統中開展研究,而陸地生態系統病毒學研究在近10年才逐漸發展。據報道每克土壤中的病毒顆粒高達107—109個[1],比細菌總數高1—2個數量級[2]。病毒在陸地生態系統中發揮著重要的生態功能[3-4],如影響宿主的死亡率和群落結構。Li等通過T4型噬菌體標記基因(g23)和細菌16S rRNA基因測序分析發現T4型噬菌體能夠影響水稻土壤細菌群落組成[5]。同時,土壤病毒可能是影響微生物死亡率和周轉速率的主要原因之一,它可裂解細胞釋放出大量的可溶性和顆粒態有機碳,促進其他微生物的轉化和利用,從而影響微生物群落結構和元素的生物地球化學循環[6-7]。此外,病毒還可通過攜帶輔助代謝基因(auxiliary metabolic genes,AMGs)影響宿主代謝,從而間接參與元素循環[4, 8],同時也為宿主提供新的功能基因以拓寬其生態位[8]。
由于土壤病毒具有龐大的數量體和潛在的生態功能,越來越多的研究人員開始關注土壤病毒。土壤病毒豐度和多樣性的研究方法主要包括熒光顯微計數法、透射電子顯微觀察、分離培養、特定標記基因分析、宏基因組學和宏病毒組學等[9-10]。宏病毒組和宏基因組是目前病毒生態學研究最重要的手段[3, 10],可以成功避免大量病毒不可培養和缺乏通用標記基因所帶來的問題,尤其是宏病毒組學可極大地增加測序序列中病毒基因組的覆蓋度。宏病毒組學研究發現在納米布沙漠土壤中,有尾噬菌體目(Caudovirales)豐度最高,其中以長尾噬菌體科(Siphoviridae)為主[11],這與南極土壤中主要病毒類群的分布一致[12]。而在中國的農田土壤中主要以單鏈DNA(ssDNA)病毒為主導,其中以微小噬菌體科(Microviridae)和環狀病毒科(Circoviridae)為主[13]。近期,Starr等首次通過宏轉錄組分析發現,植物根際與非根際土壤中的RNA病毒多樣性很高,凋落物顯著影響病毒和宿主的群落組成,而根系的生長不影響RNA病毒的群落組成[14]。
目前我國土壤宏病毒組僅有少量的研究,主要關注了農田[13, 15]、紅樹林[16],冰川[17]以及海陸交錯帶土壤[18]。不同土地利用方式會影響土壤理化性質,例如土壤pH和養分[19],以及微生物群落結構和功能[20]。但是我們對于不同土地利用方式下,病毒的群落結構和功能特征還缺乏認識。因此本研究從新疆阿克蘇沙雅縣選取兩種典型土地利用方式下的土壤(棉花地和荒漠土壤),通過宏病毒組學技術探究土壤病毒群落結構和功能特征,為進一步了解農業活動對土壤病毒分布的影響提供科學依據。
1 材料與方法
1.1 研究區域與樣品采集
采樣點位于新疆阿克蘇沙雅縣(東經82.54°、北緯41.32°),該地區屬于暖溫帶沙漠邊緣氣候區。本研究于2018年6月采集該地區棉花地土壤和荒漠土壤。棉花地種植時間約10a,與荒漠土壤之間間隔約200 m。使用梅花布點法(間隔大于3 m)布設采樣點,每個土壤樣品選取6個樣點,在每個樣點采集0—10 cm土壤樣品。土壤樣品通過冰盒運回實驗室。所有土壤通過2 mm篩去除植物殘體后,儲存于4℃冰箱用于后期土壤理化性質測定和病毒提取。
1.2 土壤理化性質測定
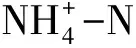
1.3 土壤病毒DNA提取及測序
分別將棉花地和荒漠土壤6個樣點的土壤混合成一個樣品,根據前期的實驗方法提取鮮土中的病毒[13],每個樣品提取1次。簡述如下:稱取過2 mm篩的鮮土500 g,與3 L的甘氨酸緩沖液(250 mM,pH=8.5)混合均勻,于150 rpm振蕩15 min。混合液于4℃ 1500 rpm條件下低速離心2 min。收集上清液,沉淀繼續添加緩沖液,重懸浮,混合均勻,并重復以上步驟兩次。利用切向流過濾系統(Tangential Flow Filter System, QuixStand, GE Healthcare Life Sciences, Pittsburgh, PA, USA)對上清液進行連續過濾,依次通過1 mm,0.45 μm和0.22 μm的濾柱,并收集透過液。再將透過液通過30 kDa的超濾柱進行濃縮并收集截留液(體積不大于100 mL)。濃縮液再經過0.22 μm的無菌過濾器過濾,重復3次以去除連續過濾過程中可能帶來的雜菌污染。再以4000 g的轉速通過30 kDa超濾離心管(Merck Millipore Ltd., Tullagreen, Ireland)進一步濃縮至體積約為1 mL。接著將病毒濃縮液用DNase I(Thermo Fisher Scientific, Lithuania, EU)在37℃孵育1 h(10 units DNase I/500 μL),以去除土壤中游離的胞外DNA。利用細菌16S rRNA基因PCR(引物27F/1492R)[21]檢測并確認沒有細菌DNA污染后進行下一步實驗。
使用病毒DNA提取試劑盒(Qiagen AllPrep PowerViral DNA/RNA extraction kit(Qiagen, Hilden, Germany))提取濃縮液中的病毒DNA。隨后根據基因組擴增試劑盒(REPLI-g Mini Kit(Qiagen, Hilden, Germany))使用說明對提取的病毒DNA進行多重置換擴增,以得到足量的病毒DNA來構建宏病毒組文庫。分別利用Nanodrop和Qubit檢測DNA的純度和濃度。再利用超聲破碎儀Covaris M220(Covaris, Woburn, MA, USA)將DNA隨機打斷成長度約為350 bp的片段,建立宏病毒組文庫后進行Ilumina HiSeq2500高通量測序。測序工作由北京新科開源基因科技有限公司完成。
1.4 宏病毒組數據分析
測序得到的原始數據通過fastp軟件,選擇默認參數[22]過濾接頭并去除低質量序列,最后得到clean reads。利用SortMeRNA v2.1去除clean reads中的核糖體序列[23],利用bbmap比對NCBI UniVec數據庫(ftp://ftp.ncbi.nlm.nih.gov/pub/UniVec, March 2017)去除可移動遺傳元件。然后利用metaSPAdes[24]對clean reads進行組裝拼接得到原始contigs。Clean reads與病毒數據庫進行BLASTx比對和物種注釋(E-Value<1e-5,score值> 50)[25],病毒數據庫綜合了NCBI non-redundant(NR)數據庫,Refseq virus 數據庫和PHAST網站上的噬菌體數據庫(截至2018年8月)[26-28]。比對得到的病毒序列使用Prodigal以默認參數進行開放閱讀框(ORFs)預測[29],并與NR和RefSeq病毒蛋白數據庫進行比對(Diamond,BLASTp,E-Value<1e-5,identity>60%)[25],最后在MEGAN 6中利用SEED數據庫對病毒進行功能分類[30]。通過Virsorter預測病毒contigs[31],contigs僅選取于具有更高可信度的categories 1,2,5。病毒基因組示意圖通過Easyfig進行可視化[32]。
1.5 序列提交
本研究中的兩個宏病毒組原始序列已經提交至NCBI Sequence Read Archive(SRA),獲取號為PRJNA599555。
2 結果與分析
2.1 土壤理化性質

表1 土壤理化性質
2.2 土壤病毒組信息
通過宏病毒組測序,從新疆棉花地和荒漠土壤樣品中分別獲得78072330和80219056條clean reads,GC含量分別為53.56%和49.45%。在棉花地和荒漠土壤宏病毒組中分別有12.34%和11.57%的reads比對上病毒數據庫。對clean reads進行拼接,在棉花地和荒漠土壤宏病毒組中分別獲得3050和2771條contigs(>1000 bp),最長的兩條contigs分別為13586 bp和17062 bp(表2)。
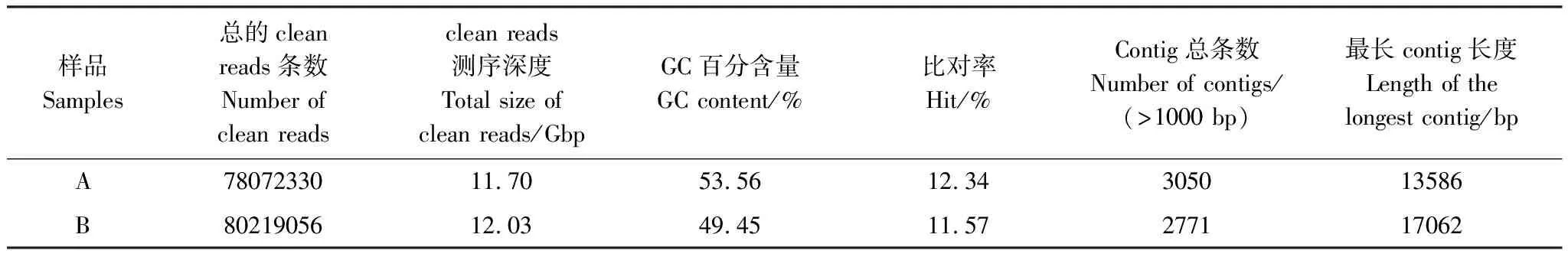
表2 病毒組信息
2.3 病毒群落結構
通過病毒數據庫注釋,新疆棉花地和荒漠土壤宏病毒組分別注釋到20個和15個病毒科和一些未分類的病毒(圖1)。在兩種土壤中,均是ssDNA病毒占絕對優勢,其中微小噬菌體科占比最高,分別為69%和78%。微小噬菌體科主要包括蘑菇狀噬菌體亞科(Gokushovirinae),分別占據棉花地和荒漠土壤宏病毒組的35%和39%。環狀病毒科在棉花地和荒漠土壤中所占比例分別為2.0%和0.7%,矮縮病毒科(Nanoviridae)在兩種土壤中所占比例分別為0.4%和0.2%。兩種土壤中均檢測到Genomoviridae、雙生病毒科(Geminiviridae)、短尾噬菌體科(Podoviridae)、肌尾噬菌體科(Myoviridae)、長尾噬菌體科、Alphasatellitidae、Smacoviridae和脫氧核糖核酸病毒科(Phycodnaviridae)等。此外在棉花地土壤中特有的病毒有花椰菜花葉病毒科(Caulimoviridae)、逆轉錄病毒科(Retroviridae)、裸露病毒科(Nudiviridae)、多分DNA病毒科(Polydnaviridae)、桿狀病毒科(Baculoviridae)和囊泡病毒科(Ascoviridae)。而乳頭瘤病毒科(Papillomaviridae)僅在荒漠土壤病毒組中檢測到。

圖1 兩個土壤病毒組物種注釋結果(基于reads注釋)
2.4 病毒功能特征
利用MEGAN的SEED數據庫對兩種土壤的病毒功能進行分類,Level 1水平的主要病毒功能占比類似,均是“Phages, Prophages, Transposable elements”和“Phages, Prophages, Transposable elements, Plasmids”占比最高(圖2)。在棉花地土壤中注釋到更多的病毒功能分類單元,除以上兩種功能外還注釋到“Amino Acids and Derivatives”、“Carbohydrates”、“Cofactors, Vitamins, Prosthetic Groups, Pigments”和“Photosynthesis”。在level 2水平上,兩個土壤一共注釋到30個病毒功能分類單元,其中“Phage capsid proteins”所占比例最高,其次是“Phage packaging machinery”和“Phage head and packaging”(圖2)。

圖2 病毒功能注釋
2.5 病毒基因組分析
通過Virsorter預測病毒序列和功能注釋,在棉花田土壤病毒組中注釋到711條病毒contigs(> 1 kb),在荒漠土壤病毒組中注釋到1113條病毒contigs(> 1 kb)。兩個土壤中的病毒contigs均主要注釋到微小噬菌體科。大于5 kb的contigs一共有147條,選擇其中最長的兩條contigs分析其功能基因組成(圖3)。Contig 1攜帶14個基因,其中包括8個病毒相關功能基因(protein affiliated to virus),5個未知功能基因(unknown function)和1個假設蛋白(hypothetical protein)。Contig 2攜帶18個基因,其中包括9個病毒相關功能基因,1個未知功能基因和8個假設蛋白(圖3)。
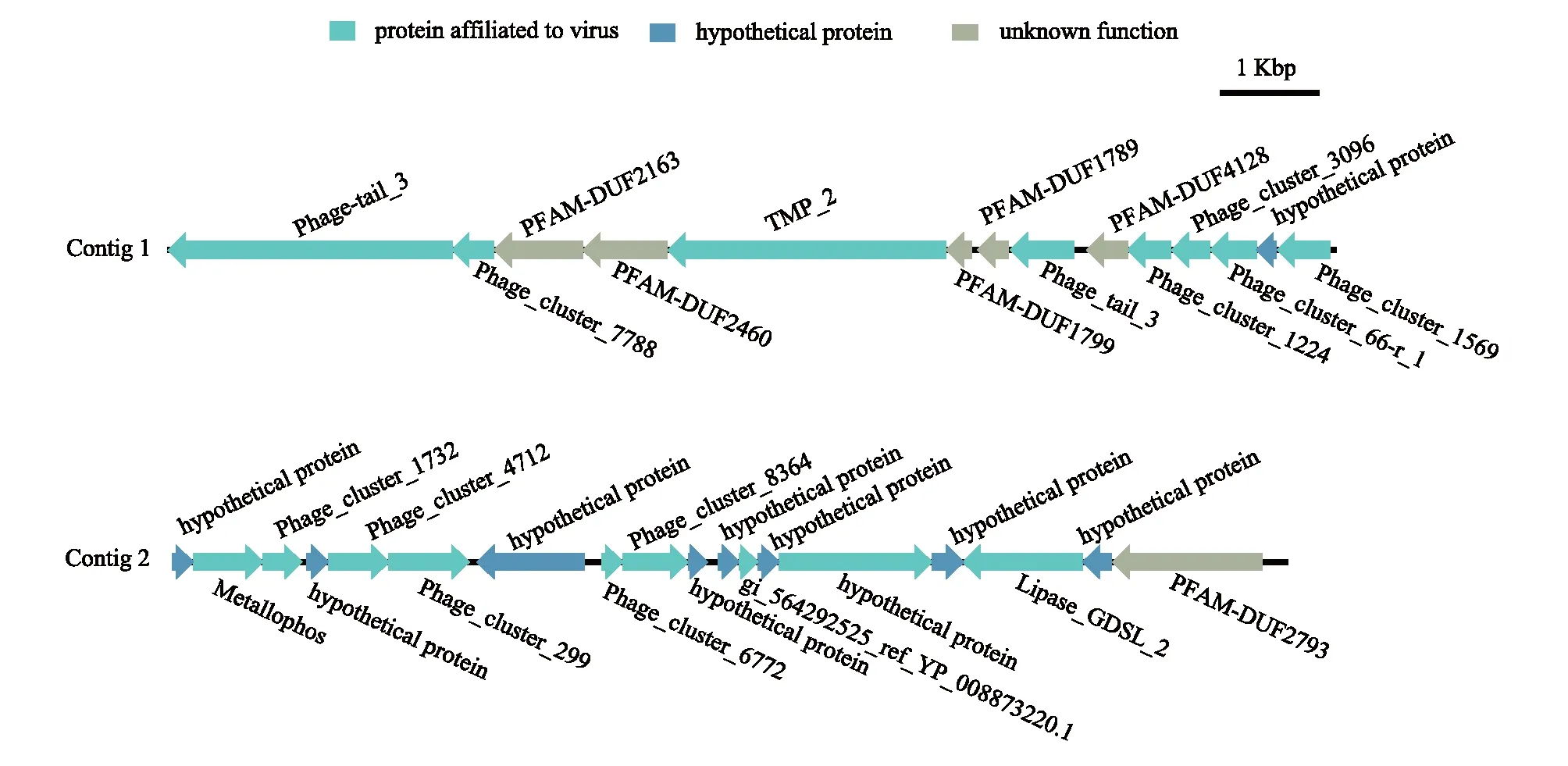
圖3 基因組示意圖
3 討論
3.1 新疆兩種土地利用方式下土壤病毒群落組成
土壤微生物在自然和人工生態系統中都扮演著十分重要的角色。標記基因和宏基因組學等研究方法極大地推動了我們對土壤微生物的認知和應用,例如合理利用微生物來提高土壤肥力、增加作物產量,增強了我們對陸地生態系統響應環境變化的理解[33]。病毒也在陸地生態系統中發揮著重要的作用,例如通過侵染微生物來調控宿主代謝、影響宿主死亡率等[34]。加州大學戴維斯分校Joanne B.Emerson副教授提出土壤病毒學是土壤微生物學研究的前沿熱點,并指出未來可以探究土壤病毒對有機質降解、碳氮元素循環、溫室氣體排放以及農業生產的影響[35],從而補充土壤微生物對生態過程的貢獻。
本研究通過宏病毒組學分析發現,新疆兩種不同土地利用方式下的土壤均富集到大量的ssDNA病毒,其中以微小噬菌體科為主。ssDNA病毒一般包括絲狀病毒科(Inoviridae)和微小噬菌體科兩個噬菌體病毒科和五個真核病毒科[36]。微小噬菌體科是具有環狀單鏈DNA基因組的二十面體的噬菌體家族,主要侵染腸桿菌、細胞內寄生菌和螺原體[37]。微小噬菌體科在感染周期中的潛伏期短、子代釋放量大[38],從而廣泛分布于環境中,如海洋、污水、沉積物和人體腸道等均有分布。2015年Brian Reavy等人通過宏病毒組學研究發現,蘇格蘭濱海土壤和農田土壤中ssDNA病毒數量占比最大,以微小噬菌體科為主(分別占84.6%和50.32%)[39]。而在紅樹林土壤中以動物病毒環狀病毒科占主導,其次是微小噬菌體科[16]。有研究指出多重置換擴增可能會偏好性地放大ssDNA病毒[40],從而導致結果中ssDNA病毒比例偏高。在棉花地和荒漠土壤中,dsDNA病毒有尾噬菌體目(包括長尾噬菌體科、肌尾噬菌體科和短尾噬菌體科)所占比例比較低。有尾噬菌體目在部分土壤樣品中也占據主導地位,例如納米布沙漠土壤[11]、美國某些農田土壤[41]以及我國新疆冰川土壤[17]。

3.2 土壤病毒功能特性
棉花地和荒漠土壤宏病毒組的主要病毒功能注釋結果相似,均主要注釋到與病毒結構相關的功能,其中“Phage capsid proteins”和“Phage packaging machinery”占比最高(圖2)。兩條全長病毒基因組序列的分析也表明可注釋的編碼基因均以病毒結構相關功能為主,未知功能蛋白仍占較高比例(圖3)。“phage capsid proteins”是病毒相關的衣殼蛋白,可作為某些特定病毒類群的標記基因,例如T4噬菌體的g23基因[44-45]和微小噬菌體科的VP1蛋白[46]。在納米布沙漠土壤病毒功能中,“phages, prophages, transposable elements, plasmids”占比最高(49%)[11]。這也與一些美國農田土壤病毒的主要功能類似[41],而部分病毒功能分類單元在農田表層和深層土壤中的相對豐度差異較大,在深層土壤中檢測到更多的噬菌體結構相關功能(如噬菌體衣殼、尾部和頸部纖維)。但在中國南方紅壤中,僅有少部分病毒功能在玉米根際與非根際土壤之間存在顯著差異[15]。此外,相對于其他土壤,棉花地和荒漠土壤病毒組功能類群較為單一[2, 41]。這也可能受到土樣樣品數量的限制,對于不同土地利用方式下土壤病毒功能特征與差異還需要進一步擴大樣品進行探索。此外,由于目前我們對病毒功能基因的分析主要依賴現有數據庫,而相關數據庫還不完善,功能注釋仍存在局限性,因此土壤病毒功能特征還有待進一步地探索和挖掘。
4 結論
本研究通過宏病毒學方法分析了新疆兩種土地利用方式下土壤病毒的群落組成和功能特征。棉花地和荒漠土壤的理化性質存在顯著差異,兩種土壤分別注釋到20個和15個病毒科,均以ssDNA病毒為優勢類群,其中以微小噬菌體科占絕對優勢。在棉花地土壤中檢測到更高比例的植物病毒(矮縮病毒科和雙生病毒科)。此外,僅在棉花地土壤中檢測到與植物相關的花椰菜花葉病毒科,與昆蟲相關的裸露病毒科、多分DNA病毒科、桿狀病毒科和囊泡病毒科。這些結果表明土壤病毒群落組成可能受到與土地利用方式相關的人為活動、土壤理化性質和動植物的影響。在兩種土壤中共獲得1824條病毒contigs,主要匹配到微小噬菌體科。用SEED數據庫對病毒功能進行注釋,棉花地和荒漠土壤病毒組的功能注釋均以病毒結構相關功能基因占比最高。本研究探索了新疆農田土壤和荒漠土壤病毒組的特征,未來研究人員還需要在更大時空尺度和基于更多樣品的土地利用方式上探究土壤病毒生態學,并進一步闡明土壤病毒對陸地生態過程的貢獻。
猜你喜歡
中華詩詞(2022年6期)2022-12-31 06:41:24
中國科技論壇(2017年7期)2017-07-25 08:49:53
財經(2017年15期)2017-07-03 22:40:49
財經(2017年2期)2017-03-10 14:35:35
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
