第三代測序技術在遺傳性耳聾基因拷貝數變異檢測的臨床應用
2021-04-25 03:56:06王秋權黃莎莎袁永一康東洋吳婕張昕戴樸
中華耳科學雜志 2021年2期
關鍵詞:檢測
王秋權 黃莎莎 袁永一 康東洋 吳婕 張昕 戴樸
中國人民解放軍總醫院耳鼻咽喉頭頸外科醫學部,國家耳鼻咽喉疾病臨床醫學研究中心,聾病教育部重點實驗室,聾病防治北京市重點實驗室(北京 100853)
拷貝數變異(copy number variations,CNVs)廣泛存在于人類基因組中,是遺傳變異的重要來源。其遺傳致病機制包括基因劑量效應、位置效應等多種分子機制[1-4]。已有研究表明,CNVs也是遺傳性耳聾的一個重要原因,占所有非綜合征性耳聾分子病因的五分之一[5-8],已成為遺傳性耳聾分子診斷的主要檢測內容之一。
過去研究人員提出的通過比較CNVs與標準參照基因之間相對值的檢測技術,如多重鏈接探針擴增技術(Multiplex Ligation-dependent Probe Amplification,MLPA),染色體微陣列分析技術(Chromosomal Microarray Analysis,CMA),下一代測序技術(Next Generation Sequencing,NGS),盡管已經大幅提高了基因組的研究能力,但在分析CNVs等大片段結構性變異方面仍存在技術局限性。近年來出現的第三代測序技術(Third-Generation Sequencing,TGS)具有超長讀長,快速,實時,高通量的優點,可以更直接有效準確地檢測CNVs,確定CNVs片段長度等參考數據,精確定位CNVs位置,找到確切斷點信息[9-11]。
本研究應用基于納米孔(Nanopore)測序技術原理的TGS平臺對1例耳聾伴有耳蝸分隔不全III型(Incomplete Partition type III,IP-III)的患者進行全基因組結構變異檢測,發現并定位了一段位于Xq21.1(chrX:81079396-84457540)區 域 且 包 含POU3F4基因的半合子缺失變異,明確了該患兒遺傳性耳聾的分子病因,為進一步遺傳咨詢及產前診斷提供了依據,現報告如下。
1 材料與方法
1.1 研究對象
先證者為一個3歲男童,父母聽力正常,非近親婚配,自然懷孕,足月生產,孕期正常。出生后雙耳聽力篩查未通過。其父母提供相關病史,家族史等信息,對其進行了相關臨床評估,診斷性聽力學檢查,包括穩態聽覺誘發電位(Auditory Steady State Potential Response,ASSR)、雙側畸變產物耳聲發射(Distortion Product Otoacoustic Emission,DPOAE)、聽性腦干反應閾值及潛伏期(Auditory Brainstem Response,ABR),及顳骨CT等。對患兒進行全外顯子組測序后未檢測到與臨床表型相關的具有可能臨床意義的變異,僅通過XHMM CNV軟件預測到一段位于Xq21.1區域的半合子缺失變異,由于全外顯子組測序的局限性及軟件預測存在假陽性,并不能直接輔助臨床診斷,因此我們對該患兒進行了TGS測序。該研究得到中國人民解放軍總醫院倫理委員會的批準(倫理編號:S2106-120-01)。
1.2 研究方法
1.2.1 基因組DNA提取及文庫制備
簽署知情同意書后,抽取先證者及父母外周血各2mL(EDTA抗凝),采用Genomic DNA提取試劑盒(Qiagen,Germany)并根據生產廠商提供的標準操作流程進行樣品基因組DNA抽提,所得DNA使用NanoDrop One UV-Vis spectrophotometer(Thermo,USA)檢測DNA純度。樣本質檢合格后,使用BluePippin全自動核酸回收儀(Sage Science,USA)將大片段進行切膠回收,對DNA進行損傷修復,純化后在DNA片段兩端進行末端修復并進行加“A”反應,使用LSK109連接試劑盒(Oxford Nanopore Technologies,UK)中的接頭進行連接反應,最后用Qubit 3.0 Fluorometer(Invitrogen,USA)對建好的DNA文庫進行精確定量檢測。
1.2.2 對先證者進行Nanopore測序
建庫完成后將一定濃度和體積的DNA文庫加入到1個Flow cell中,并將Flow cell轉移到PromethION(Oxford Nanopore Technologies,UK)進行實時納米孔單分子測序。
1.2.3 生物信息學分析
對Nanopore測序下機數據進行統計并進行過濾。對獲取的數據用NGMLR-Sniffles流程進行結構變異檢測。用ANNOVAR進行結構變異注釋。應用1000 genome phase3、DGV gold standard CNV、dbVar nstd37和DESPICHER結構變異注釋。根據疾病的表型調研OMIM、HPO和Clinvar等數據庫,閱讀相關文獻,確定疾病相關基因。
1.2.4 結果驗證
對患兒父母親分別進行Sanger測序驗證,驗證CNVs的準確斷點位置信息。
2.資源配置是改革發展的基石。供給側改革就是對有限的資源配置進行優化,進而有高質量的產出,繼續教育供給側改革的資源配置首先要有制度的保障。現階段,各地政府陸續出臺了地方性的終身教育條例。在這些終身教育條例中,經費保障被寫入條例中來。比如廣東省的經費保障是常住人口每人每年不低于2元,上海市經費保障稍高些,達到了每人每年不低于5元的保障。有了必要的制度、經費、人員保障,繼續教育項目的開發等要緊扣當地社會經濟支柱產業的需求。比如,廣東省將先進制造業等作為十大支柱產業,那么繼續教育就要統籌好有限的資源,圍繞這些支柱產業開發優質教育資源,為當地社會經濟發展提供智力支持。
2 結果
2.1 臨床結果
先證者ASSR顯示雙側極重度感音神經性耳聾(圖1a);雙耳鼓室導抗圖均為A型;雙側DPOAE在正常范圍內均未引出;ABR閾值及潛伏期示雙耳分別給與click聲刺激,雙耳在90dB(nHL)下見V波分化,均未見CM波分化。
顳骨CT顯示雙側耳蝸頂轉與中轉融合,體積較小,蝸軸缺如,內聽道與耳蝸相通,雙側內聽道擴大,按照Sennaroglu分類[12],為 IP-III型,雙側前庭略擴大,雙側面神經管迷路段與鼓室段夾角增大(圖 1b)。

圖1 a.ASSR檢查示示雙側極重度感音神經性耳聾;b.顳骨CT示雙側內耳畸形,蝸軸缺失并與內聽道相通,為耳蝸分隔不全III型(IP-III)。Fig.1 a.ASSR examination showed bilateral severe sensorineural hearing loss;b.CT of the temporal bone showed bilateral inner ear malformation,cochlear axis missing and communicating with the internal auditory canal,which belonged to incomplete partition type III(IP-III).
2.2 基因檢測結果
2.2.1 生物信息學分析
檢測到先證者攜帶一段位于Xq21.1(chrX:81079396-84457540)區域的3.38Mb左右的半合子缺失變異,由于該變異缺失片段較長,無法同時得到正常DNA片段和發生變異DNA片段,故分別在缺失變異后的融合位點兩側(chrX:81078896-81079395,chrX:84457541-844578,圖 2c中F-R引物)和斷點兩側(chrX:84457254-84458046,圖2c中f-r引物)設計引物擴增該家系樣品后進行瓊脂糖電泳實驗。瓊脂糖電泳顯示患兒只擴增出突變型序列條帶,其父親只擴增出野生型序列條帶,其受檢者母親既擴增出突變型序列條帶也擴增出野生型序列條帶(圖2d)。一代驗證結果表明該缺失變異來自于患兒母親(圖2e)。

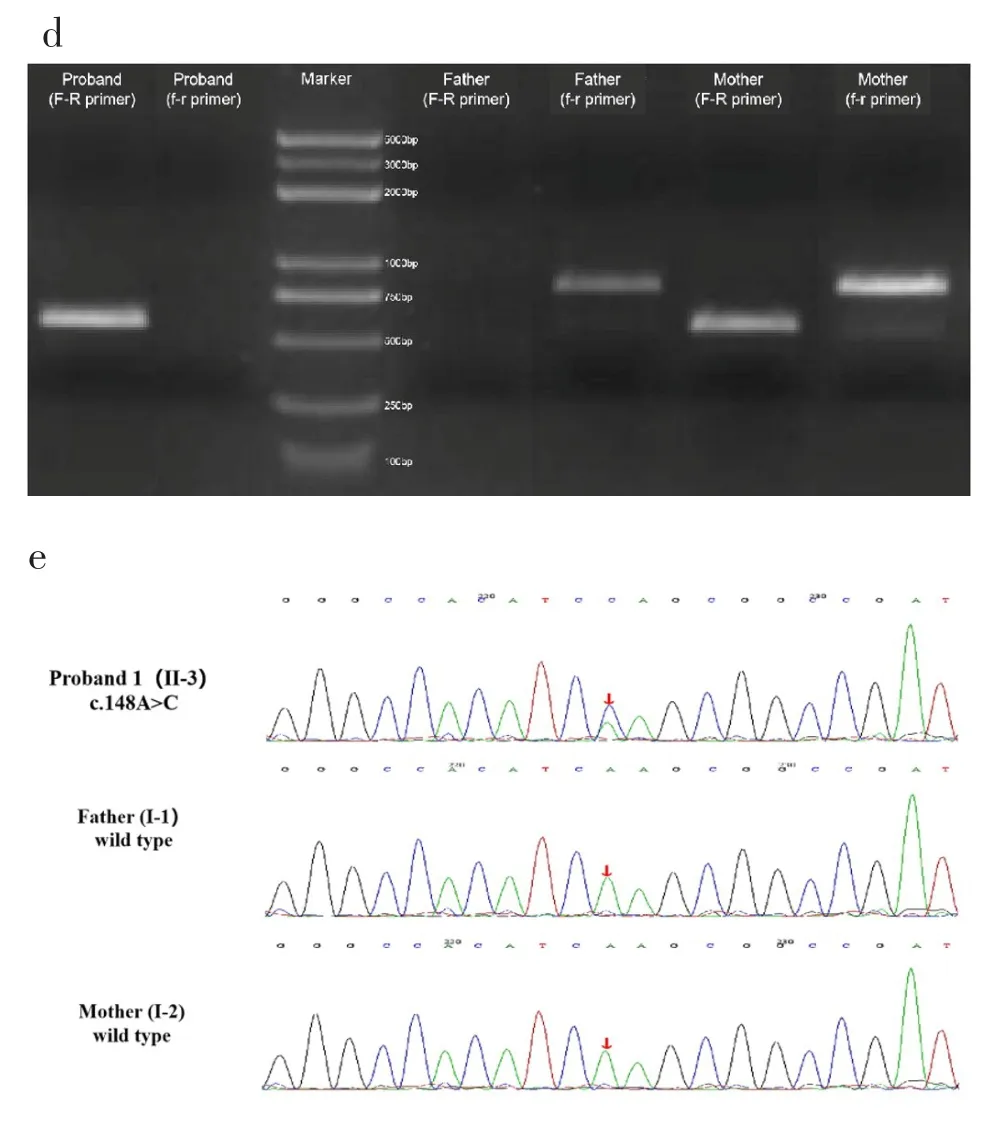
圖2 a.家系圖譜;b.Nanopore測序結果顯示先證者攜帶一段位于Xq21.1(chrX:81079396-84457540)區域的3.38Mb左右缺失變異;c.缺失變異后的融合位點兩側和斷點兩側設計引物;d.瓊脂糖電泳顯示受檢者只擴增出突變型序列條帶,其父親只擴增出野生型序列條帶,其母親既擴增出突變型序列條帶也擴增出野生型序列條帶;e.Sanger測序驗證結果表明該缺失變異來自于母親。Fig.2 a.The pedigre;b.Nanopore sequencing results showed that the proband carry a 3.38 Mb deletion variant in theXq21.1(chrX:81079396-84457540);c.Primers were designed on both sides of fusion site and breakpoints after deletion of mutation;d.Agarose electrophoresis showed that the proband amplified only mutant sequence bands,father amplified only wild-type sequence bands,and mother amplified both mutant and wild-type sequence bands;e.Sanger sequencing validation results indicated that the deletion variant was derived from the mother.
2.2.2 致病性分析
該缺失變異覆蓋了 APOOL、CYLC1、HDX、POU3F4、RPS6KA6、SATL1基因。除SATL1基因未被收錄,APOOL、CYLC1、HDX、RPS6KA6基因在OMIM數據庫中未收錄相關疾病,因此排除上述基因致病性。此外,并未發現其他致病性單核苷酸變異。
該段缺失CNVs覆蓋的POU3F4基因(OMIM:#300039)在OMIM數據庫中明確記錄為X-連鎖耳聾2型(OMIM:#304400;DFNX2)的致病基因,該變異使POU3F4基因編碼序列完全缺失,影響編碼蛋白正常功能(PVS1)。通過查詢DGV數據庫和gnomADSVs數據庫中均未見該變異的收錄(PM2)。但在DESPICHER、ISCA及ClinVar數據庫及相關文獻中均有覆蓋該變異區段或大部分重疊的缺失變異,并被定義為致病性或疑似致病性變異,相關臨床表型均有聽力障礙(PM3)。基于父母親驗證結果及以上證據,根據ACMG指南[13],判定該變異為致病性變異(PVS1+PM2+PM3),POU3F4基因為致病基因。
3 討論
在多個耳聾患者中通過NGS等測序技術檢出僅包含POU3F4基因全部序列的缺失變異,均位于本研究檢測到的變異區間范圍內,且本患者的臨床特征與其中報道的耳聾男性患者表型一致[19-23]。該變異導致POU3F4基因序列全部丟失,使功能性POU3F4蛋白缺乏,從而破壞中耳和內耳結構的正常發育,導致聽力障礙。
此外,也有多例Xq21區域的大片段缺失使POU3F4基因與相鄰基因組成綜合征型聽力障礙的報道,具體表型取決于其CNVs大小和致病基因含量,主要為DFNX2、注意力缺陷、智力障礙和脈絡膜出血等[24-28]。盡管本研究病例的缺失范圍也覆蓋其他基因,但該患者并未出現其他特殊的表型,且覆蓋的其他基因目前并無明確的致病性證據,因此排除該患者為綜合征型聽力障礙。然而,并不能排除其他基因編碼的蛋白質與已知致病基因可能存在相互作用,并產生與觀察到的表型相關的功能變化,這需要我們進一步通過全基因組分析來鑒定。
POU3F4基因位于X染色體上的基因沙漠中,該區域并未覆蓋有許多重要功能基因,且富含高度保守的非編碼區,這可能導致POU3F4基因或相應區域較頻繁發生CNVs[29]。而要闡明POU3F4基因CNVs發生的確切機制,需進一步對CNVs斷點周圍重復序列等分析,因此,斷點的精確定位對探索POU3F4基因CNVs的發生機制并評估其功能影響至關重要。傳統的檢測方法對于CNVs斷點的精確定位均有一定的局限性,需面對如斷點所在區域富含復雜重復元件,高GC率或存在假基因、測序讀長短,不能準確映射回正確基因組位置等問題[30,31]。本研究應用的Oxford Nanopor平臺是一種基于納米孔測序的第三代單分子基因測序技術。該技術不僅具有高通量特點,同時可以得到CNVs兩端序列,即使斷點出現在高度重復和復雜區域中,也可進行更精確的映射與基因組比較;而該技術的長讀長測序,克服了傳統檢測技術短讀長的諸多局限,使獲得更大無間隙的全長基因組并對其進行結構序列分析成為可能[32,33]。
明確先證者CNVs致病性可對先證者母親再次妊娠前準確的遺傳咨詢和產前診斷提供有力支持。本研究中先證者母親再次妊娠,在孕19周通過采集羊水檢查與超聲檢查對胎兒進行了產前診斷及性別鑒定,結果顯示胎兒性別為女性,其攜帶該片段的雜合變異。對于X連鎖隱性遺傳疾病來說,單個致病性變異的女性攜帶者并不會發展為患者,只有當男性基因型為半合子,女性基因型為純合子或攜帶復合雜合致病性變異(如反式)時才會導致耳聾的發生。因此,胎兒同其母親一樣均為該缺失變異的攜帶者,并不會重復其哥哥的表型。出生后經隨訪,該女嬰聽力正常。
4 結論
本研究通過TGS檢測及Sanger測序驗證的方法明確了一名伴有內耳畸形的遺傳性耳聾患者的分子病因,并對先證者母親再次妊娠胎兒進行了遺傳咨詢及產前診斷。本研究證明TGS用于遺傳性耳聾基因中CNVs的臨床應用是可行的,但仍面臨著諸多問題,如單讀長的錯誤率偏高,需重復測序以糾錯;生物信息學分析軟件不夠豐富、數據積累少;價格成本高等,其有效性和準確度仍需大量數據的評估和驗證。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
