固體在液相中吸附熱力學參數計算介紹
2021-04-09 11:15:54王偉濤陳香李楊百勤
大學化學 2021年2期
王偉濤,陳香李,楊百勤
陜西科技大學化學與化工學院,西安 710021
吸附現象在實際生活和生產中廣泛存在。吸附是指在固相-氣相、固相-液相、液相-液相等體系中,氣相或者液相組分在表面上發生富集的現象[1]。固體在液相中吸附應用十分廣泛,如染色、脫色、廢水處理、水凈化、液相組分的分離和提純等方面。
固體表面能夠發生吸附現象主要由于固體表面的不均勻性。固體表面并不是簡單的體相中止,也不是體相結構的簡單延續。從原子水平看,固體的表面并不是光滑的,表面的原子存在很多種可能的結構環境(圖1)。所以,固體表面原子處于化學鍵不飽和狀態,具有較高的能量,即熱力學上處于不穩定狀態,必然會自發地吸附氣體或溶液中的分子,降低其表面自由能。因此,吸附過程是自發的自由能減小過程。
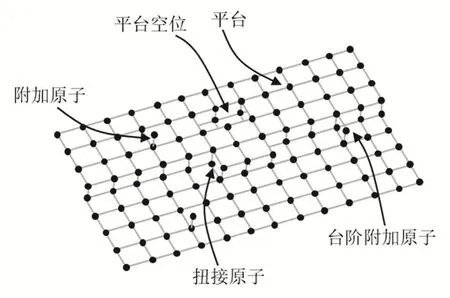
圖1 固體表面原子情況示意圖[2]
被吸附的物質稱為吸附質(adsorbate),具有吸附作用的物質稱為吸附劑(adsorbent)。吸附作用力有化學鍵、氫鍵、酸堿、共軛π-π作用、靜電力、范德華力等作用力。吸附熱力學參數可以判斷吸附過程能否自發進行,吸附自發進行的熱力學趨勢大小;通過吸附熱力學可以進一步分析吸附質、吸附劑、溶劑等吸附體系和條件對吸附過程的影響[3]。用熱力學方法處理吸附平衡問題時,一般需要熱力學參數來對其進行分析。因此,研究固-液吸附時需要以吸附熱力學參數來分析其吸附熱力學,即吸附吉布斯自由能變(ΔG)、吸附焓變(ΔH)、吸附熵變(ΔS)。
盡管吸附熱力學已經成為研究吸附現象的必要手段,但是沒有專門的文獻介紹吸附過程熱力學參數的計算方法,致使初學者在研究吸附熱力學性質時需要查閱大量的文獻。另外,在眾多文獻報道中吸附熱力學參數計算方法不同,并且文獻中一般只是直接引用所使用的計算公式,這對初學者來說會帶來很大的困惑。因此,本文通過介紹不同方法計算吸附熱力學參數的推導,并通過舉例介紹吸附熱力學參數的計算和應用。
1 固-液界面吸附過程化學勢
固-液界面的吸附可以看作特殊的相變過程,吸附平衡是一種特殊形式的相平衡[4,5]。因此,可以利用相平衡原理來處理吸附平衡。液相中某組分i在固-液界面上發生吸附,吸附過程是組分i從液相中遷移到固-液界面上:
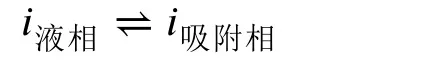
達到吸附平衡時,根據組分i在液相中和界面上的化學勢相等,可以建立關系式。
液相中組分i的化學勢:

吸附在固體表面的組分i化學勢:
式中,fi和fads,i分別為組分i在液相和在吸附相中的活度因子;ci和cads,i分為組分i在液相和吸附相中的濃度;ai和aads,i分為組分i在液相和吸附相中的活度。

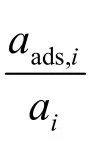

2 固-液界面吸附過程ΔG計算
從式(7)中可以看出,要計算吉布斯函數變化ΔG?,需要先計算吸附平衡常數K。K值是計算吸附過程熱力學函數變化的關鍵,但是不同文獻中對K的計算方法各不相同。因此,我們有必要厘清K的計算。
2.1 極限法獲得K計算ΔG

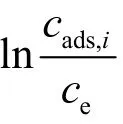

式中,ρ1為溶劑的密度(g·mL?1),M1為溶劑的分子量(g·mol?1),A1為溶劑分子的橫截面積(cm2·molecule?1),s為吸附劑的表面積(cm2·g?1),x/m 為比吸附量(μg·g?1),M2為吸附的組分 i分子量(g·mol?1),式中得到 cads,i的單位為 mol·L?1。這樣就可以求出 K。

2.2 利用平衡濃度獲得K計算ΔG
直接使用溶液中溶質的濃度變化量和平衡濃度的比值計算K[11],即

式中,c0為溶液中溶質的初始濃度,ce為平衡濃度。利用此式可以簡單迅速地計算出K。該式是以(c0? ce)代替了吸附相中組分i的濃度cads,i;只有在稀溶液下才可以近似認為活度因子為1,和式(8)一致,因為K的定義為活度之比,否則也需要利用極限法計算。在溶液中吸附,(c0? ce)和cads,i存在一定的差異;當吸附劑對溶劑的吸附量大于對溶質的吸附時,此時(c0? ce)為負值,從而導致K為負值,而無法利用式(7)計算ΔG;而實際上,cads,i只可能為正值。因而,該方法計算并不是很準確,僅僅可以粗略估算吸附熱力學參數的變化趨勢。
直接使用平衡吸附量和平衡濃度的比值計算K[12],即

式中,qe為吸附平衡時吸附劑對吸附質的表觀吸附量(mol·g?1),ce為平衡濃度(mol·L?1)。該式是在式(10)的基礎上,認為在極低稀溶液時,在吸附相上吸附量非常小,組分i在吸附相的濃度數值上約等于吸附量。因而,該式僅限于無限稀溶液下的計算;同時,也存在式(10)的缺陷:qe和K有可能也為負值,而無法計算ΔG。另外,式(11)計算存在的另一個問題是K有量綱(L·g?1)。為此,有研究者提出[3],K值應該乘以溶劑的濃度(g·L?1),即可得到較為合適的K。
2.3 利用等溫吸附模型中的常數計算
固體在液相中的吸附體系遠比固-氣界面的吸附復雜。固體在液相吸附時,固體表面存在對溶質和溶劑的競爭吸附,而這種競爭是溶質-溶質、溶劑-溶劑、溶劑-溶質、溶質-吸附劑、溶劑-吸附劑之間作用的綜合結果。目前,液相吸附的理論多是沿用氣體吸附的理論模型,雖然液相吸附的實驗結果在形式上能用氣體吸附模型處理,但其也只是經驗性的。
根據式(7),只要求出吸附過程的平衡常數K,就可以求出 ΔG。通過擬合等溫吸附過程的吸附模型,直接以等溫吸附模型方程式中的吸附常數代替K來計算ΔG[13]。常見的等溫吸附模型如表1所示。文獻中報道最多的是Langmuir和Freundlich方程,多數吸附等溫模型符合這兩個模型中的一個。當符合Langmuir方程或Freundlich方程時,直接用方程中的KL或KF代替K[14]計算ΔG。當符合Herry方程,以Herry方程中的常數KH計算時,有KH=qe/ce,與式(11)一致。利用等溫吸附模型中的常數計算吸附熱力學參數,可以同時獲得符合的等溫吸附模型和吸熱的熱力學參數。需要指出的是,不同吸附模型得到的ΔG之間不具有可比性,畢竟K和等溫模型方程式中的吸附常數不同;但是得到的計算結果并不影響其吸附熱力學上的趨勢規律。

表1 不同的吸附等溫模型及其方程表達式
2.4 利用Freundlich公式參數法計算ΔG
Gibbs吸附等溫式可以寫成[15]:

式中,ΔG?為吸附自由能變(J),a為溶質在溶液中的活度,N為吸附的溶質摩爾數(mol)。為了獲得單位質量的吸附吉布斯自由能變化,式(13)可以變化為:

式中,ΔG?為吸附自由能變(J·g?1),q為吸附量(mol·g?1)。當濃度較低時,可以用濃度或摩爾分數代替活度[15]。用摩爾分數代替活度時,式(14)變為:

式中,x為溶液中溶質的摩爾分數。將Freundlich方程式中的吸附量q = KFx1/n帶入式(15)中,可以得到:

積分后,得

對ΔG′′除以q,可以得到每摩爾溶質吸附的吉布斯自由能變(J·mol-1),即

這是一個和吸附量無關的吉布斯自由能變的計算公式,式中n為Freundlich方程式中的常數,T為溫度(K)。一般來說,對于符合Freundlich吸附模型的等溫吸附來說,在計算Gibbs自由能變時,只要獲得Freundlich吸附模型方程式中的常數n,就可以直接用ΔG = ?nRT即可。
在研究吸附過程吉布斯自由能變時,需要根據實際情況選擇合適的計算方法。采用不同方法計算的吸附自由能變數值可能不同,但是其變化規律是一致的。因此,如果要比較不同吸附體系的熱力學函數ΔG,需要采用同一種計算方法。一般來說,物理吸附的自由能變化絕對值小于化學自由能變絕對值,物理吸附自由能變一般為?20 - 0 kJ·mol?1,化學吸附的自由能變一般為?400 - ?80 kJ·mol?1。因此,可以通過ΔG的數值來判斷吸附過程是物理吸附還是化學吸附過程。
3 固-液界面吸附過程ΔH計算
3.1 利用平衡常數K求ΔH
吸附焓變(或者吸附熱)可以通過Van’t Hoff方程計算[16]:

式中,K為吸附平衡常數。設ΔH不是溫度的函數,可以對其進行積分,得到:

通過不同的溫度下的平衡常數K和溫度T,可以求出吸附焓變ΔH。式(21)也可以由ΔG = ΔH ?TΔS直接得到。將ΔG = ?RTlnK帶入,可得到:

由式(23)可以知道,式(21)中的常數C為ΔS/R。利用式(21)或者式(23),以不同溫度下的lnK對1/T作圖,得到一條直線,根據直線斜率求出ΔH。文獻也有此式的變化形式[17]:

式(21)、式(23)、式(24)本質上是一致的。
3.2 利用平衡濃度ce求ΔH
在稀溶液中,活度可以近似等于摩爾分數;同時,假定在吸附量一定時,吸附劑上吸附的溶質和溶劑的比例是常數;可以得到平衡常數和平衡時溶液中溶質摩爾分數成反比,和平衡時溶液中溶質的濃度成反比[15],即:

將其帶入Van’t Hoff方程的定積分式(120)中,則Van’t Hoff方程變為:

式中,ce,1和ce,2分別表示不同溫度下達到吸附平衡時的濃度。
將式(25)帶入Van’t Hoff方程的不定積分式(21)中,可以得到:

通過lnce對1/T作圖,得到一條直線,根據直線的斜率可以求出ΔH。該式和文獻中報道的利用Clausius-Clapeyron方程求解ΔH的方程式形式一致[18]。
除此之外,ΔH還可以利用Gibbs-Helmholtz方程計算[10]:

只要求出了ΔG,可以方便的求出ΔH。
通過吸附熱的大小可以粗略判斷吸附作用力類型[19,20]。吸附作用力有化學鍵、氫鍵、電子供體受體作用、共軛π-π作用、靜電力、范德華力等作用力。當吸附作用力為范德華力時,吸附熱在4-10 kJ·mol?1;吸附作用力為氫鍵時,吸附熱為 2-40 kJ·mol?1;作用力為配位基交換、偶極間作用力時,吸附熱分別為~40和2-29 kJ·mol?1;當吸附作用力為化學鍵時,吸附熱大于60 kJ·mol?1。但是,吸附過程的吸附熱是吸附質和吸附劑之間多種作用力的綜合作用結果,通過吸附熱判斷作用力的類型只是一個粗略的估計。另外,可以用吸附焓變來區分吸附過程是化學吸附或物理吸附。一般認為,焓變的絕對值< 40 kJ·mol?1時,為物理吸附;焓變的絕對值在50-200 kJ·mol?1時,則為化學吸附。如果吸附焓變為負值,則表示吸附過程是放熱的,升高溫度將不利于吸附;反之如果吸附焓為正值,則表示吸附過程是吸熱的,升高溫度將利于吸附。
4 固-液界面吸附過程ΔS計算
得到ΔG后,ΔS可以根據下式進行計算[21]:

在求 ΔH時,可以利用式(23),以 lnK對1/T作圖,利用直線的截距 ΔS/R可以計算出ΔS。另外,用已經求出的ΔG和ΔH,通過Gibbs-Helmholtz方程直接計算:

在固體自氣相吸附,氣體由三維的氣相轉移到二維表面相時,其自由度降低,氣體的吸附過程伴隨的是熵的減少,熵變為負值;所以,氣相物理吸附的驅動力一般是焓變而不是熵變。但是在液相吸附時,溶質和溶劑都會發生吸附,溶質的吸附是熵減少的過程;溶質的吸附必然伴隨溶劑的脫附,溶劑分子的脫附是熵增過程[10]。所以,吸附過程熵變應該是二者共同作用的結果。如果吸附過程僅僅是物理吸附,同時溶質分子面積大于溶劑分子面積時,意味著吸附一個溶質分子可能會有大于一個的溶劑分子脫附;所以,溶質吸附引起的熵減可能小于溶劑分子脫附引起的熵增,其吸附過程的熵變可能是正值。因此,吸附過程的熵是增大還是減小,和溶質和溶劑分子的相對大小有關。
5 應用舉例
5.1 活性炭自水溶液中吸附對二甲苯
KC-8活性炭自水溶液中吸附對二甲苯,通過對其吸附等溫線擬合,發現其符合Freundlich吸附模型,因此可以根據式(18)計算ΔG,根據式(27)計算ΔH,根據式(30)計算ΔS,得到的吸附熱力學參數如表2所示[22]。該吸附過程的ΔG <0且其值在?20 - 0 kJ·mol?1范圍內,表明該吸附過程是一個自發的物理吸附過程。該吸附過程的ΔH> 0,表明該吸附過程是吸熱過程;根據ΔH值的大小并結合ΔG值可以判斷,該吸附作用力不可能為化學鍵作用力。吸附熵變ΔS> 0,因此可以推斷該吸附過程伴隨著溶劑的脫附過程。當活性炭吸附對二甲苯時,其自由度減少,對應的熵減少;當溶劑分子(水)脫附時,熵值增大。當吸附一個對二甲苯分子時,就會有多個溶劑水分子脫附,因而該吸附過程總熵變大于0。

表2 在313.15 K下KC-8活性炭自水溶液中吸附對二甲苯的熱力學參數[22]
5.2 硅膠對同系物的吸附
硅膠自四氯化碳中吸附脂肪醇同系物,計算得到的ΔG如表3所示[23]。從表中可以看出隨著脂肪醇碳鏈的增長,對應的ΔG增大,表明在非極性溶劑中極性吸附劑對極性吸附質的作用較強。隨著碳鏈長度增加,每一個CH2官能團對ΔG的貢獻值并不是一個常數。這表明脂肪醇并不是以“平躺式”在界面吸附。在稀溶液中,單官能團極性有機物是以其極性基團吸附在硅膠表面,非極性的疏水基團以一定的方式留在非極性溶液中[5]。

表3 硅膠從正脂肪醇的四氯化碳溶液中吸附的ΔG [23]
硅膠從環己烷中吸附苯、萘、蒽、菲的熱力學參數如表4所示[21]。硅膠對苯、萘、蒽、菲的吸附ΔG差別不大。這是由于這四種物質的結構相似,在硅膠表面的吸附都是由π電子對固體表面的作用而吸附,吸附時都是“平躺”在界面上的。
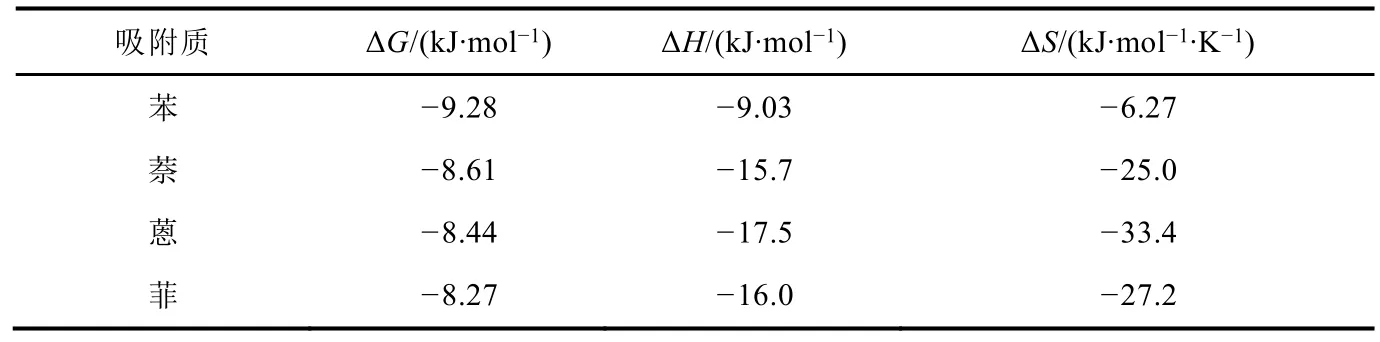
表4 硅膠自環己烷中吸附苯、萘、蒽、菲的熱力學參數[5]
6 結語
吸附過程的熱力學參數對研究吸附劑吸附性質具有重要的指導意義。吸附熱力學參數的計算方法有多種,不同的計算方法得到計算結果可能不同。因此,不同計算方法得到計算結果不具有可比性;但是不同計算方法不改變吸附熱力學參數的變化規律。通過獲得的熱力學參數可以判斷吸附過程是物理吸附或化學吸附,也可以判斷吸附質和吸附劑之間作用強弱及粗略判斷吸附作用力的類型;同時,可以通過吸附的熱力學參數之間的比較可以推斷溶質分子結構對吸附的影響及吸附方式。
