稠油油藏儲層冷采用活性分子的性能評價與應用*
2021-04-09 03:15:46楊祖國高秋英
油田化學 2021年1期
關鍵詞:質量
楊祖國,高秋英,任 波,路 熙
(1.中國石油化工股份有限公司西北油田分公司,新疆烏魯木齊 830011;2.中國石化股份有限公司石油勘探開發研究院,北京 100083;3.中國石化縫洞型油藏提高采收率重點實驗室,新疆烏魯木齊 830011)
我國稠油資源極為豐富,已在渤海灣盆地、松遼盆地、南襄盆地、準噶爾盆地、二連盆地等12個大中型含油盆地和凹陷發現70 多個稠油油田,稠油(包括瀝青)儲量在80×108t以上。中國石化稠油資源集中在勝利、中原和河南油田,目前主要以蒸汽吞吐為開采方法并輔助少量常規冷采。稠油黏度高,牛頓流體性質差,導致儲層流動阻力大,對于絕大多數稠油油田,除了蒸汽吞吐和蒸汽驅等熱采技術外,常規冷采的采收率非常低,小于原始儲量的5%。然而,蒸汽吞吐過高的措施費用在目前低油價下難以實現效益開采。此外,高輪次吞吐后期,汽竄嚴重,含水驟升,熱采效率下降明顯。“十二五”稠油熱采比“十一五”動用儲量減少5×107t,上產難度加大。雖然常規冷采對儲層傷害較小,但其產量過低,更無法維持稠油的長期正常生產。隨著原油黏度的進一步增加,原油在儲層中甚至失去可流動性,常規冷采已無法實現正常生產,特超稠油、深層稠油、敏感性稠油和薄層稠油總計1.3 億噸儲量處于難動用開發狀態。
面對蒸汽吞吐成本高,后期開發效益下降,常規冷采效率過低等問題,化學降黏冷采成為解決該類問題的有效方法之一[1—7]。但化學降黏冷采技術亦面臨著巨大的困難:室內降黏效果非常顯著,現場增油效果很不穩定。稠油作為假塑性流體,儲層條件下流動性較差,隨著黏度增加塑性增強,愈難以與水形成混相,僅以降低界面張力為目的的乳化活性分子溶液難以滲入稠油內部令其分散,更無法實現依托水相作為流動相的原油降黏[8—11]。為突破此技術瓶頸,必須先削弱稠油內部重質組分之間的相互作用,將活性分子溶液引入稠油內部,以此分散油相達到降黏的目的[12—15]。本文以丙烯酰胺、丙烯酸鈉、4-苯基-1-丁烯、丙烯酰胺-2-甲基丙磺酸為原料、四甲基乙二胺為催化劑制備了活性分子共聚物(AAPA),并對其性能進行了評價。通過優化措施工藝以及礦場實驗與效果分析,形成了針對邊底水薄層特稠油油藏的化學降黏冷采技術。
1 實驗部分
1.1 材料與儀器
丙烯酰胺(AM)、丙烯酸鈉(AA)、4-苯基-1-丁烯(PB)、丙烯酰胺-2-甲基丙磺酸(AMPS)、N,N'-二甲基乙二胺(DEMED)、Na2CO3、異丙醇、過硫酸胺、偶氮異丁腈(AIBN)均為分析純,煤油,上海阿拉丁試劑公司;鹽酸、NaOH、MgCl2、CaCl2、NaCl,化學純,國藥集團化學試劑有限公司;P14 井稠油,地面脫氣黏度3.91×104mPa·s(50℃),瀝青質含量20.86%。
LP1002 型電子天平,常熟市衡器廠;CH702 型恒溫攪拌水浴槽,鞏義市予華儀器有限責任公司;IKA-T18 型高速乳化器,德國IKA 公司;DHG-9070A型電熱鼓風干燥箱,上海一恒科學儀器有限公司;DV-Ⅱ+Pro黏度計,美國Brookfield公司。
1.2 實驗方法
(1)活性分子共聚物(AAPA)的制備
活性分子共聚物(AAPA)的合成路線如圖1 所示。將300 g去離子水和100 g煤油加入1500 mL三口圓底燒瓶中,放入磁子攪拌,然后加入60 g AM、10 g AA、20 g PB 和5 g AMPS,攪拌至完全溶解并形成白色乳液。隨后加入催化劑0.15 g DEMED,攪拌均勻后加入Na2CO3將pH 值調至8。加熱升溫至70℃,通入氮氣除氧30 min,然后加入5 g 異丙醇、0.15 g過硫酸胺和0.15 g AIBN,反應至溶液變黏、磁子停止轉動時,關閉攪拌并停止通入氮氣,反應8 h。取出燒瓶中的凝膠塊,剪碎后置于烘箱中80℃加熱脫水6 h,研磨造粒至80~100目(0.180~0.150 mm),反應收率為92.6%,數均分子量為30×104~100×104。

圖1 活性分子共聚物(AAPA)的合成路線
(2)性能測試
動態攪拌條件下的降黏率(V):參照中國石化企業標準Q/SH1020 1519—2016《稠油活性分子溶液通用技術條件》,稱取一定量的脫水原油,在80℃的恒溫水浴中預熱30 min。用去離子水分別配制100~1500 mg/L的活性分子溶液,作為測試降黏劑體系。按不同的油劑質量比加入活性分子溶液,用乳化器在500 r/min 下攪拌油劑混合體系3 min,在50℃、60 r/min 下用黏度計測定其黏度,按式(1)計算動態降黏率V。

式中:μ0—加入活性分子前的原油黏度,mPa·s;μ1—加入活性分子后的原油黏度,mPa·s。
靜態條件下的降黏率(m):稱取一定質量(m0≥25 g)的脫水原油,在80℃的恒溫水浴中預熱30 min。按不同的油劑質量比加入活性分子溶液,油劑混合體系在恒溫水浴中靜置一段時間后,將試管傾斜135°,稱取倒出的稠油質量(m1),按式(2)計算靜態降黏率m。
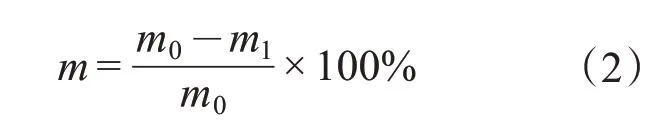
2 結果與討論
2.1 油劑質量比對降黏效果的影響
配制1000 mg/L 的AAPA 水溶液,分別按油劑質量比7∶3、6∶4、5∶5、4∶6、3∶7將原油和AAPA溶液混合,80℃加熱30 min后測定降黏率V和m,結果如圖2所示。隨著AAPA溶液比例的增加,V和m的變化趨勢基本一致,降黏效果不斷提升。然而,攪拌剪切條件下的降黏率V高于靜置條件下的降黏率m。這是由于攪拌可令稠油與降黏劑的接觸更充分,利于提高降黏效果。當油劑比為1∶0時(即無降黏劑加入的條件下),分別在50、80℃下測定原油的黏度μ0、μ1,按式(1)計算的V僅為85.1%;同時由50、80℃下可倒出的原油質量m0和m1,按式(2)計算的m僅為66.7%。溫度升高增強了分子間熱運動,進而降低了重質組份分子間作用力,令黏度出現不同程度下降。V比m高了18.4%,說明剪切可提高降黏率。當油劑比=7∶3時,受降黏劑的影響,與油劑比=1∶0 時僅靠溫度降黏相比,V和m分別提高10%和20.4%,但因降黏劑用量比例過低,降黏效果相對較差;當油劑比=5∶5 時,降黏明顯增加,V=99.4%,m=93.2%;當油劑比<5∶5 時,降黏率隨著降黏劑比例增加而提高,但增幅逐漸減緩;與油劑比為4∶6相比,油劑比為3∶7的V和m僅提高0.5%和3.9%。按照相同的質量比3∶7,將降黏劑替換為水后,降黏率與純油的情況下基本一致,說明水無法與該黏度下的原油形成乳液,無任何降黏作用。綜合考慮性價比,油劑最佳質量比為1∶1,此時稠油黏度降為332.4 mPa·s,具備良好的流動性。
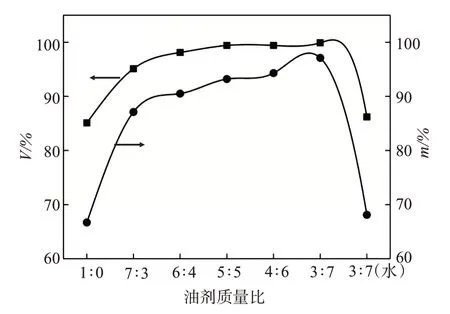
圖2 油劑質量比對原油降黏率的影響
2.2 AAPA溶液質量濃度對降黏效果的影響
配制質量濃度分別為0~1500 mg/L 的AAPA水溶液。按照油劑質量比1∶1,在20 g 稠油油樣中加入20 g AAPA 水溶液,在80℃下加熱30 min 后測定降黏率,結果如圖3 所示。兩種降黏率均隨著降黏劑濃度的增加而增加,但攪拌條件下的V仍高于靜置條件下的m。由于儲層條件下稠油黏度高、流動差,基本處于靜止或低速滲流狀態,攪拌的測試方法與實際情況相差過大,因此后續的實驗均采用靜置降黏的方法進行測試。當AAPA質量濃度低于400 mg/L時,降黏率m<75%,降黏后的稠油黏度為1221.8 mPa·s,此時稠油處于不可流動狀態,即該濃度下的活性分子溶液是無效的;當AAPA 質量濃度處于500~1000 mg/L 時,降黏效果隨之增加,質量濃度為1000 mg/L時的m=93.2%,降黏效果最佳;當濃度繼續增加時,由于聚合物水溶液本身的黏度過高,即便稠油黏度下降,也會造成儲層整體出液困難,因此AAPA溶液適宜的質量濃度為1000 mg/L。
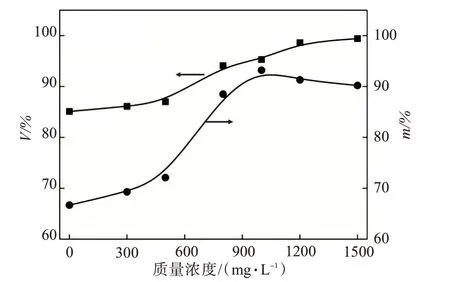
圖3 AAPA溶液質量濃度對原油降黏率的影響
2.3 酸堿、Ca2+、Mg2+對降黏效果的影響
配制1000 mg/L 的AAPA 水溶液,按油劑質量比1∶1將20 g稠油與20 gAAPA溶液混合,分別用鹽酸和NaOH 將pH 值調至1~14,在80℃下加熱30 min 后測定靜態降黏率m,結果如圖4 所示。當pH<4時,AAPA在強酸性條件下基本失活,油劑立刻出現分層,即酸性條件下可令降黏乳化后的稠油迅速破乳,避免對地面集輸及水處理產生影響。當pH>10時,AAPA在堿性條件下可將特稠油乳化分散成極小的油滴分散在水相中,進一步提升降黏率。因此,現場配液水酸值不能過低,適度提高pH值可增強AAPA的降黏活性。
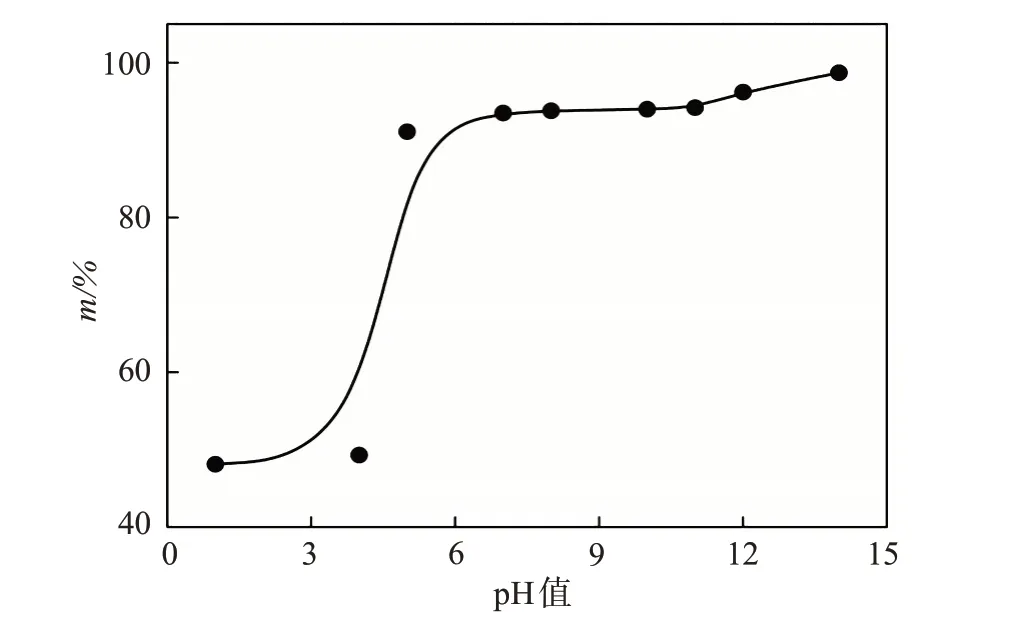
圖4 pH值對原油靜態降黏率的影響
分別配制3 g/L MgCl2溶液、10 g/L CaCl2溶液和飽和NaCl溶液,隨后加入AAPA配成100 g 1 g/L活性分子溶液。按照油劑質量比1∶1,向3個燒杯中分別加入100 g 稠油,在80℃下加熱30 min 后測定靜態降黏率m。隨后,將上述3 個燒杯降至室溫(25℃)放置4 d,3 g/L的MgCl2溶液中因MgCl2導致聚合物絮凝而出現大量沉淀,油劑分層,上層稠油黏度基本不變;10 g/L 的CaCl2溶液中因CaCl2水溶液密度遠高于油相,因此稠油基本位于上層,無沉淀析出,油層仍具有一定的流動性,仍保持降黏狀態;飽和NaCl溶液中油劑分層,稠油流動性良好,降黏率未受飽和NaCl的影響。再次在80℃下加熱30 min,10 g/L的CaCl2溶液和飽和NaCl溶液中的AAPA 仍具有降黏效果,但CaCl2溶液中稠油室溫靜置后的聚并速度明顯快于飽和NaCl溶液,3 g/L MgCl2溶液中的AAPA基本失活。
分別測量不同Ca2+、Mg2+濃度下的靜態降黏率m,通過擬合得到AAPA 耐Ca2+、Mg2+的極限有效濃度,結果如圖5 所示。圖5(a)中隨著Ca2+質量濃度的增加,降黏率降幅較小,Ca2+質量濃度為10 g/L時的m=88%,儲層溫度下原油降黏后的黏度為586.5 mPa·s,具備良好的流動性,故AAPA 的耐Ca2+量可達10 g/L。由于單體AMPS 中的磺酸基團可與Ca2+通過靜電作用緊密結合,可有效避免Ca2+引起的高分子絮凝現象。圖5(b)中當Mg2+質量濃度超過1.4 g/L 時,降黏效果迅速下降,根據擬合結果,當Mg2+質量濃度>3 g/L時,AAPA基本失去降黏能力。由于AAPA 中存在AA 單體,其中的—COO-官能團與Mg2+會生成—COOMg沉淀,導致聚合物AAPA從水相中絮凝沉淀失去降黏活性,燒杯中出現明顯的白色沉淀。
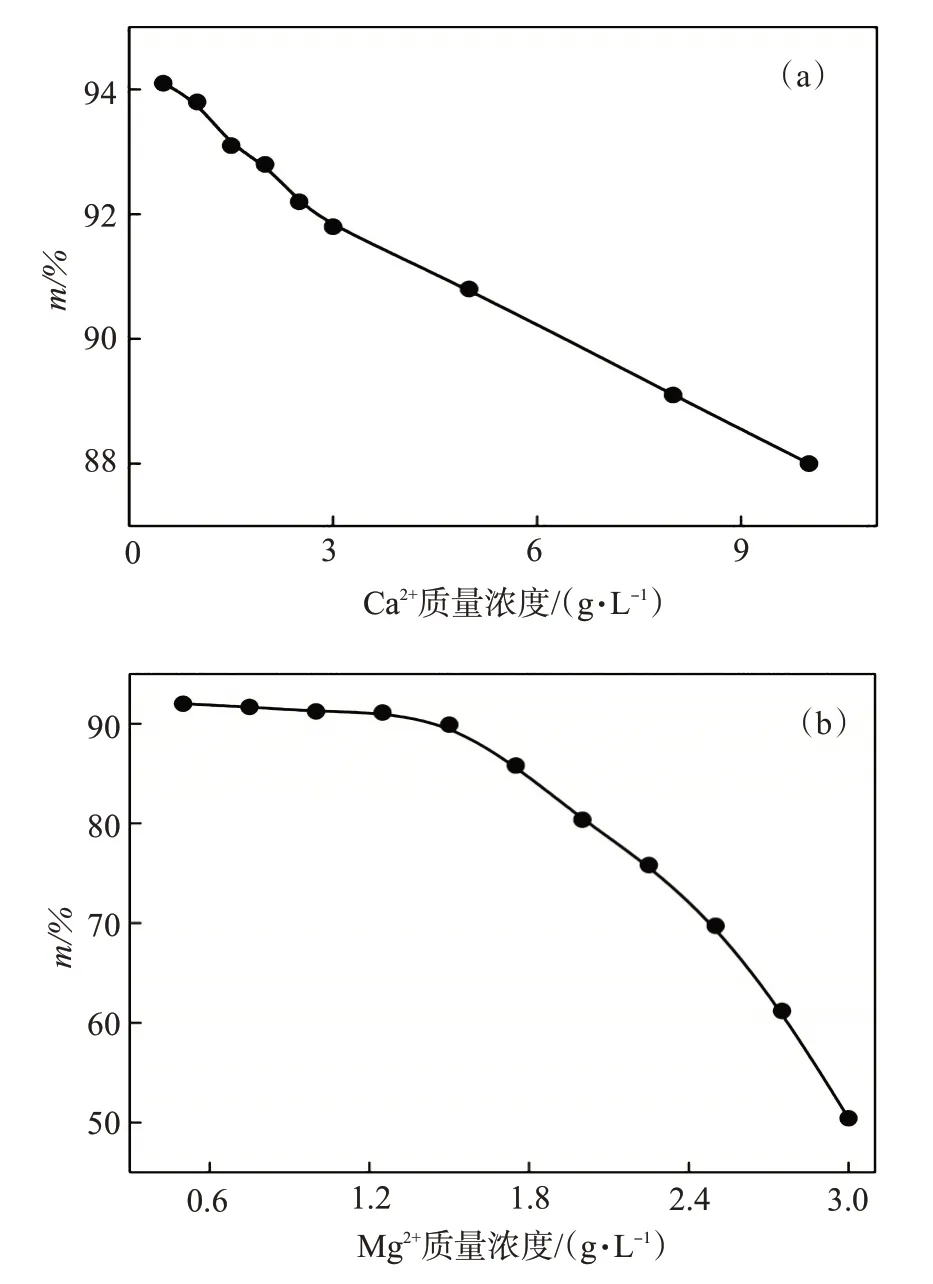
圖5 Ca2+、Mg2+質量濃度對靜態降黏率的影響
2.4 溫度對降黏效果的影響
在20 g稠油中加入20 g 1 g/L的AAPA溶液,分別在50~80℃下靜置加熱不同時間后,測定靜態降黏率m,結果如表1所示。加熱2 h后,50℃下AAPA溶液與稠油依然處于明顯的分層狀態,此時稠油的黏度為39100 mPa·s,降黏劑滲入效率低,難以達到降黏效果。持續加熱5 d后,降黏劑與稠油混合,降黏率m=80.1%。60、70℃下的AAPA 溶液與稠油則發生不同程度的融合,加熱4 h 后才實現充分互溶。僅80℃下AAPA 溶液與稠油在加熱30 min 后形成油劑混溶乳液,實現降黏。因此,溫度低于80℃時,需要相應的增加加熱時間以達到充分降黏的目的。溫度為50℃時的m=80.1%,降黏后的稠油黏度為7780.9 mPa·s,盡管稠油處于難流動狀態,但卻依然具有一定的降黏性能。當溫度為70、80℃時,AAPA 的降黏效果接近,m分別為93.1%和93.9%。在80℃降黏后,冷卻至35℃時的m仍可保持在90.5%,說明AAPA 具有良好的低溫抗聚并性能。進一步從35℃冷卻至25℃,靜置2 h 后稠油再次聚并形成油塊,盡管AAPA 仍保持對稠油的降黏效果(m=90.1%),但室溫下稠油的原始黏度高達2.4×105mPa·s,以致此時稠油的黏度為23760 mPa·s,流動困難。提高溫度至100、180℃時,靜置加熱5 min 后稠油基本處于稀油狀態,迅速測得的黏度均低于100 mPa·s,冷卻至室溫的m=91.2%,可見AAPA 在180℃下具有良好的降黏性能。對于P14 井,儲層溫度80℃,井口溫度40℃(>35℃),即AAPA降黏自儲層至井筒舉升到地面是可行的。
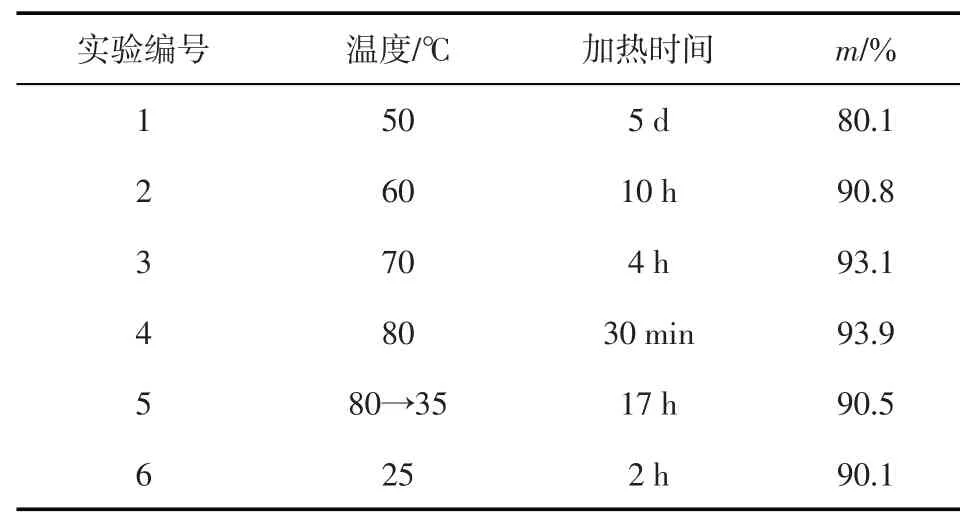
表1 溫度對原油靜態降黏率的影響
綜上所述,針對50℃下黏度<50 Pa·s的脫氣脫水稠油,活性高分子AAPA 的質量濃度為1 g/L,油劑質量比為1∶1,可實現靜置降黏率>90%;最低有效降黏溫度50℃,最高降黏溫度180oC;耐Ca2+濃度≤10 g/L,耐Mg2+濃度<3 g/L;pH<4時降黏乳化后的稠油可快速破乳,pH >10時可進一步提高AAPA的降黏活性。
2.5 降黏機理
如圖6所示,根據相似相容原理,AAPA通過PB單體中的苯環整體嵌入瀝青質的層間結構中,以此將AA與AMPS兩個極性單體引入瀝青質片層結構之間,增加了瀝青質-瀝青質之間的距離,降低了分子間作用力,破壞瀝青質結構中苯環π-π鍵的受力方向,令重質組份分子之間產生各向異性,繼而降低稠油黏度。隨后,由于AMPS 完全電離后的磺酸根基團具有極強的親水性,可誘導水分子大量進入稠油內部,且防止進入稠油內部的水分子形成油包水的反相乳液。原油內部重質組份間相互作用力被削弱,外加大量水包油乳液對其隔離,原油黏度隨之下降,且無法大量聚并導致黏度反彈。單體AM可通過提高AAPA的分子量,增加水溶液的黏度,在注入過程中減少指進,令AAPA 中的功能團與原油充分接觸,提高降黏效率。
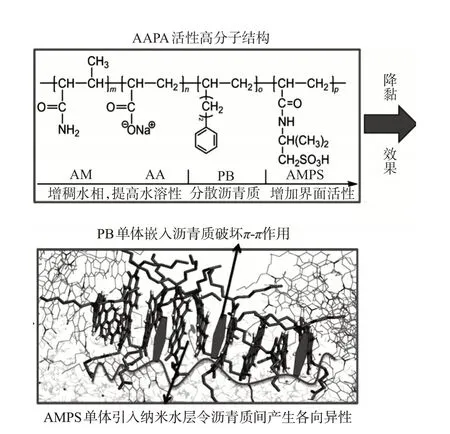
圖6 AAPA對稠油的降黏機理示意圖
2.6 AAPA儲層降黏冷采礦場應用
2.6.1 工藝技術
對于直斜井,采用三段塞濃度注入法:第一段塞,液量Q×(20%~30%),加量30~50 g/L,注入排量10~20 m3/h,對于采出程度<5%的儲層,近井區域原油動用差,低排量注入高濃度降黏劑,可令活性分子溶液充分與原油接觸,實現快速降黏,避免注劑高壓,對于含水>50%的儲層,可有效避免后續注劑被過度稀釋造成降黏劑浪費;第二段塞,液量Q×(20%~50%),加量10~30 g/L,注入排量40~60 m3/h,高排量注入可令降黏劑向遠端持續推進,避免形成稠油環導致儲層堵塞;第三段塞,液量Q×(10%~30%),加量5~10 g/L,注入排量20~40 m3/h,降低注入排量并降低注入濃度,進一步提升第二段塞高速注入下產生的注劑空間內稠油的流度,提高儲層出液能力;施工結束后,套管安裝壓力表,壓力<1 MPa,即開井生產。
對于水平井,采用四段塞濃度注入法:第一段塞,液量Q×(10%~20%),加量30~50 g/L,注入排量10~20 m3/h,避免注劑高壓,或提高近井積液的降黏劑濃度;第二段塞,液量Q×(20%~60%),加量5~10 g/L,注入排量30~50 m3/h,高排量注入可以增加注劑對B 靶點未動用區域的波及效率,提高水平段的整體動用率;第三段塞,液量Q×(10%~30%),加量10~30 g/L,注入排量10~20 m3/h,低排量注入高濃度降黏劑以維持套管區域的降黏劑濃度,提高近井儲層的滲透率,增加后續生產的出液能力;第四段塞,液量Q×(10%~20%),加量5~10 g/L,注入排量40~60 m3/h,以最低有效濃度作為頂替液,增加前置注劑的作用范圍,令更多稠油的流度得到提升;施工結束后,套管安裝壓力表,壓力<1 MPa,即開井生產。
2.6.2 礦場應用
根據AAPA 的降黏性能與優化的工藝設計,首先對P14井儲層降黏后,冷采峰值產量達到7.5 t/d,持續12 d。措施效果持續14 個月,累計產液1.18×104t,產油2348 t,增油1025 t。按照措施成本=措施費用/周期累油計算(運行成本相同,故不計算在內),AAPA儲層降黏冷采的措施成本比蒸汽吞吐的最低措施成本降低約27%,比蒸汽吞吐三輪最高產量的平均成本降低約49.5%。
基于P14 井成功實現措施增油,繼續開展了3口井的礦場試驗。沾18-5-側平12井為強底水特稠油儲層,注汽極易造成底水錐進。應用AAPA 儲層降黏冷采增油效果顯著,液量增加4 倍,產油提高3倍,措施后峰值產量高達13.7 t/d,持續40 d,接近目前該區塊熱采的最高單井沾18-P25 井的峰值產量(14 t/d)。周期持續增油13 個月,累計產油高達3933 t,措施效益明顯。同時,由于措施后采出稠油黏度顯著下降,井筒舉升良好,于措施后2個月內停止井筒摻水舉升,節約了一定的生產運行成本。陳371-平29井為邊底水特稠油油藏,經歷六輪蒸汽吞吐后含水高達98.5%,產量僅0.3 t/d。措施后通過降低近井稠油黏度,有效提高水平井整體出液效率,將液量提高47.6 t/d,產量提高到4.3 t/d,同時高輪次后期的高含水也得到有效控制,降至90.9%。
P29-75 井作為礦場實驗唯一的直井,屬于新層上返,但油層有效厚度僅1.4 m,且存在強邊底水。上返后,第一輪注汽過程中由于出現套損,導致含水高達100%,無法正常生產。套管補漏后,采取AAPA 儲層降黏,產量恢復到1.6 t/d。與水平井相比,直井的出液區域僅存在于射孔段,相應的,降黏劑也只能經射孔段進入地層,持續地注劑會對近井油層形成“反驅”效應,令原油遠離井筒區域,以致后續注劑觸油幾率下降,儲層降黏效果受限。相比之下,水平井更適合采用儲層降黏冷采,增油效果更好。
可通過儲層降黏有效實現近井解堵,遠端洗油的效果,措施后生產中集“儲層-井筒-集輸”降黏于一體,更利于整體提高產量。此外,AAPA儲層降黏冷采效果持續周期至少12個月,水平井周期平均累采超過1500 t,直井周期累采約500 t,單次措施成本較蒸汽吞吐降低明顯。
3 結論
以丙烯酰胺(AM)、丙烯酸鈉(AA)、4-苯基-1-丁烯(PB)、丙烯酰胺-2-甲基丙磺酸(AMPS)為原料、四甲基乙二胺為催化劑制備了活性分子共聚物(AAPA)。AAPA 的有效降黏溫度范圍為50~180℃;耐Ca2+質量濃度≤10 g/L,耐Mg2+質量濃度<3 g/L;pH<4 時降黏乳化后的稠油可快速破乳,pH>10 時可進一步提高AAPA 的降黏活性。1 g/L的AAPA與原油以質量比1∶1混合后的降黏效果最佳,可使黏度(50℃)<50 Pa·s的脫氣脫水稠油的靜置降黏率>90%。AAPA 具有耐高礦化度、易于破乳以及降黏溫度適用區間寬泛的特性。在勝利油田實施化學降黏冷采措施后,稠油黏度降低,提液增產效果明顯,經濟效益顯著。該技術是邊底水薄層特稠油油藏高輪次注汽后期穩產、提產有效的接替技術之一,對國內油區過億噸特、超稠油未動用及難動用儲量的開發有著重要的意義。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
