原發脊柱Rosai-Dorfman病的影像學表現
2021-03-22 02:20:34何小華李向東
中國骨科臨床與基礎研究雜志 2021年3期
關鍵詞:信號
陳 耿,王 婕,何小華,李向東,王 蔚
Rosai-Dorfman 病(Rosai-Dorfman disease,RDD)又稱竇組織細胞增生伴巨淋巴結病(sinus histiocytosis with massive lymphadenopathy,SHML),是一種罕見的良性組織增生癥[1],主要位于淋巴結內,淋巴結外好發于皮膚、上呼吸道和骨,其次為胃腸道、下呼吸道、口腔及軟組織等,原發于脊柱的RDD 罕見[2]。本研究回顧性分析9 例經病理確診的原發淋巴結外脊柱RDD 患者的臨床資料,分析其CT、MRI 征象特點及鑒別診斷要點,以期提高對原發淋巴結外脊柱RDD 的認識,為臨床診療提供參考依據。
1 資料與方法
1.1 一般資料
收集2012年3月至2020年6月經手術病理證實的9 例原發淋巴結外脊柱RDD 患者,其中男6例,女3例,年齡19~65歲。臨床表現主要為脊髓受壓和/或神經根壓迫引起的頸部或胸背部疼痛、肢體乏力等癥狀,持續且無明顯緩解,不伴發熱及淋巴結腫大等全身癥狀。全部病例均行MRI平掃及增強掃描,7例行CT平掃。
1.2 影像學檢查和圖像分析
患者取仰臥位。CT掃描采用西門子雙源CT機,管電壓120 kV,管電流100~360 mA,重建層厚5 mm。MRI 掃描采用西門子sonata 1.5T 和GE HDXt 3.0T超導型MRI成像系統,脊柱線圈。具體參數:矢狀位T2WI序列(FSE序列,TR=2 320 ms,TE=110 ms,層厚3 mm,層間距1 mm,FOV 240 mm×240 mm/320 mm×320 mm),矢狀位T1WI 序列(FSE 序列/T1FLAIR 序列,TR=500 ms/600 ms,TE=20 ms,層厚3 mm,層間距1 mm,FOV 240 mm×240 mm/320 mm×320 mm),橫軸位T2WI 序列(FSE序列,TR=3 000 ms,TE=90 ms,層厚3 mm,層間距0.5 mm,FOV 200 mm×200 mm)。經肘靜脈注射對比劑泛影葡胺(劑量0.1 mmol/kg 體質量)進行增強掃描,包括橫軸位、冠狀位及矢狀位T1WI 壓脂序列(各參數同平掃T1WI),掃描層面及層數與平掃一致。
由2 名放射科高年資主治以上醫師采用盲法獨立閱片,分析相應圖像,明確病變位置、形態、密度/信號、強化方式及鄰近骨質改變等。意見不同時協商達成一致。
2 結果
2.1 病變部位
2例位于頸椎,4例位于胸椎,1例同時累及頸椎、胸椎及顱底,2例頸椎及胸椎均有病灶,所有病灶位于椎體或椎管內髓外硬膜外/下,累及相應硬脊膜。
2.2 影像學表現
2.2.1 CT 表現 平掃呈等密度,CT 值44~85 HU。3 例累及相應椎體/附件(2 例位于寰樞椎),CT 表現為骨質溶骨性改變,骨皮質不完整,其中1例內見點狀殘存骨嵴(呈高密度),未見骨膜反應,周圍未見明顯軟組織腫塊影;6例位于椎管內髓外硬膜外/下,CT對病變的顯示不如MRI。
2.2.2 MRI表現 以周圍肌肉為參考,T1WI序列呈等信號,T2WI 序列呈等/稍低信號,增強掃描呈均勻明顯強化,但強化程度大部分弱于相鄰脊膜,累及相應硬脊膜并沿脊膜生長,可見“長尾巴”征,部分病例相應水平脊髓受壓、變性/不變性;2例病例累及椎旁軟組織。所有病例未見明顯包膜,內部未見壞死、液化、出血、鈣化表現。
2.3 手術方式、大體及病理學結果
根據病變部位、性質、侵犯范圍選擇手術方式。4例(位于胸椎)行胸椎后路腫物完全切除術,3 例(1 例頸椎、胸椎及顱底均有病灶,2 例頸椎及胸椎均有病灶)行胸椎后路腫物部分切除術,2 例(位于寰樞椎)行頸椎后路椎體復位內固定術+自體髂骨植骨術(僅取部分組織活檢)。
術中肉眼可見病變呈灰白色/黃棕色/白色魚肉狀,與周圍軟組織分界不清,質地中等堅韌,血供豐富。活檢組織病理學結果提示纖維結締組織增生,可見組織細胞、淋巴細胞、漿細胞和少許中性粒細胞浸潤,其中組織細胞大部分體積較大,胞漿豐富且透明,內有較大空泡狀核,局部可見吞噬淋巴細胞和漿細胞的組織細胞;免疫組織化學染色:特異性表達S-100和CD68,不表達CD1a。
2.4 術后隨訪
本組4 例病灶完全切除,隨訪時間1 年6 個月至3年不等,復查MRI均未見復發征象。
3例患者術后殘留病灶,其中2例給予輔助放射治療,1 例放療3 個月后疼痛癥狀未緩解,復查MRI提示復發,再次行手術切除,術后疼痛仍未消失,予以糖皮質激素治療6個月,隨訪1年6個月病灶基本消失(圖1),疼痛稍好轉;另1例隨訪2年未見復發征象。1 例未做進一步治療,隨訪2 年6 個月復查MRI 未有明確病變進展,臨床癥狀亦未見加重。
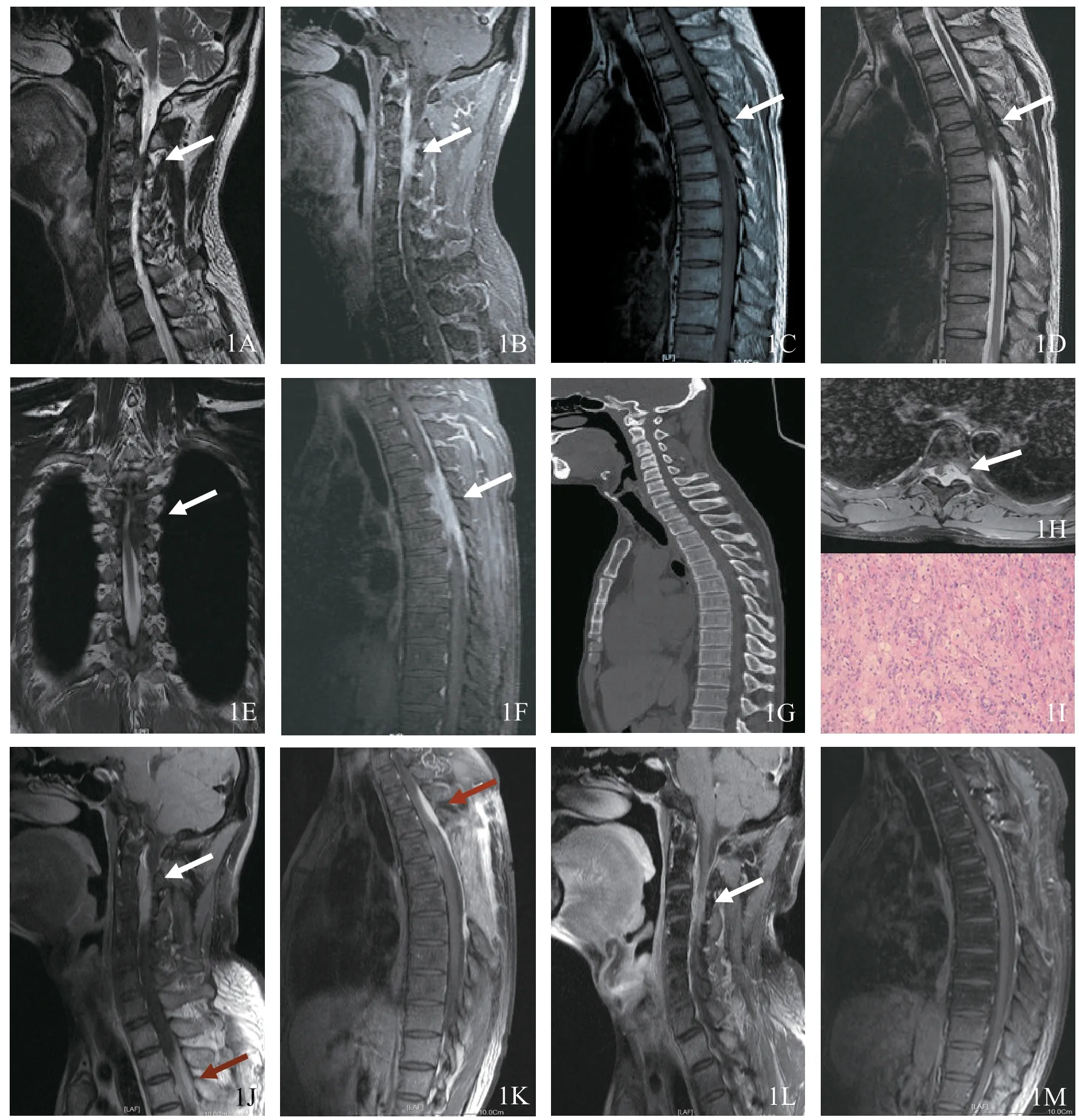
圖1 原發脊柱Rosai-Dorfman病MRI表現(男,39歲)1A 頸椎矢狀位T2WI,可見C3~C4水平椎管內髓外右側稍低信號腫塊(白色箭頭所示)1B 增強掃描T1WI+FS 可見病變明顯強化,相應周圍脊膜可見強化,有長尾巴征表現 1C 胸椎矢狀位T1WI,T4~T7水平椎管內髓外可見等信號病變 1D 矢狀位T2WI病變呈等、稍低混雜信號 1E 冠狀位T2WI示脊髓略受壓1F 增強掃描病變明顯均勻強化,相應周圍脊膜可見強化,有長尾巴征表現 1G 頸胸椎CT矢狀位重建示椎體骨質未有明顯異常改變 1H 胸椎CT增強掃描橫軸位示病變位于髓外向椎旁生長 1I 活檢組織病理學圖片示纖維結締組織間散在組織細胞吞噬淋巴細胞、漿細胞(蘇木精-伊紅染色,×200)1J,1K 椎管內腫物部分切除術+頸胸椎病變部位放療后3個月,頸椎、胸椎增強掃描T1WI+FS示C3~C4水平椎管內髓外右側仍見腫塊,T4~T7水平椎管內髓外腫塊大部分切除,T2~T3水平椎管內可見新發腫塊(紅色箭頭所示)1L,1M T2~T3椎管內腫物切除術+激素治療,術后18 個月復查頸椎、胸椎增強掃描T1WI+FS示C3~C4水平椎管內髓外右側腫塊縮小,胸椎腫塊基本消失
2 例患者僅取部分病灶活檢,未做任何治療,分別隨訪1 年、2 年6 個月,復查CT 及MRI 結果,提示骨質受侵情況較前好轉,骨質局部出現硬化表現,臨床癥狀未見明顯加重。
3 討論
RDD 是由Rosai 和Dorfman[1]于1969 年首次詳細報道并命名的一種罕見良性組織增生癥,2016 年WHO 中樞神經系統腫瘤分類將該病列入組織細胞腫瘤范疇[3]。根據病變發生的部位,RDD可分為淋巴結型、結外孤立型、混合型(同時累及淋巴結和淋巴結外器官)3種亞型[4],其中以淋巴結型為主,原發于脊柱的罕見。目前RDD的發病原因仍不明確,一般認為可能與病毒感染、免疫功能障礙有關[4]。
3.1 臨床特點及組織病理學特點
原發脊柱RDD 可位于椎體、髓內或髓外,好發于30~50歲中年人,男性稍多見,患者多因脊髓和(或)神經根的壓迫癥狀就診,臨床表現為反復頸部/胸背部疼痛,痙攣癱或截癱,不伴淋巴結受累、發熱、血沉加快等全身癥狀。大部分患者肌力及肌張力正常,部分患者手掌及手指感覺減退。
RDD確診需要依靠組織病理學檢查[4-5],其組織病理特點與淋巴結內病變相似[6-7],結節狀分布的結構特點及“明暗”相間的分布特征是診斷RDD 病的重要線索。其中“明區”內組織細胞大部分體積較大,胞漿豐富且透明,內有較大的空泡狀核,可見小核仁,局部可見特征性“伸入現象”[7];“暗區”內主要是浸潤的炎癥細胞。免疫組化特異性組織細胞表達S-100 和CD68,不表達CD1a,有助于確診。
3.2 影像學特征
復習相關文獻[8-10],結合本研究結果,原發脊柱RDD 的影像學表現主要具有以下特點:①病灶可單發或多發,多發者可呈跳躍性,未見明確包膜。②CT 具有較高的空間分辨率,對骨質的顯示較好,病灶呈等密度。③MRI 具有良好的軟組織分辨率及多參數成像特點,對于顯示腫塊范圍及與周圍結構毗鄰關系有較大優勢;病灶T1WI序列呈等信號,T2WI 呈稍低/等信號,增強掃描呈均勻明顯強化,提示病變部位血供豐富;病灶累及相應硬脊膜,可見“長尾巴”征,手術操作時病灶可能與周圍組織無法分離。④病灶內部密度/信號均勻,壞死、液化、出血或鈣化罕見。⑤病變可侵蝕周圍骨質或發生于椎體,但一般為溶骨性骨質改變,可伴有反應性骨質硬化,部分內見粗大骨嵴,提示良性病變,有自限性傾向。
3.3 鑒別診斷
淋巴結外原發脊柱RDD 需與脊膜瘤、淋巴瘤、朗格漢斯組織細胞增生癥、結核、非特異性炎癥或肉芽腫等進行鑒別。
3.3.1 脊膜瘤 起源于蛛網膜內皮細胞或硬脊膜的纖維細胞[11],一般自硬膜下呈寬基底向椎管內生長,邊界清晰,伴有脊膜尾征(一般長度較RDD短),增強掃描強化程度與硬脊膜一致,且通常較RDD明顯。脊膜瘤往往引起骨質受壓、邊緣硬化,很少向椎管外生長;RDD則附著于脊膜上生長,邊界不清,可向椎管外生長。
3.3.2 淋巴瘤 以累及椎體為主,單發或多發,自椎體向椎管內生長,骨質破壞為溶骨性[12],不會出現硬化和粗大骨嵴,不出現長尾巴征,可伴有椎管內髓外硬膜外腫塊及椎旁軟組織腫塊,多位于胸段,范圍較長,一般呈包鞘狀環繞脊髓硬膜囊生長[13],常伴有全身癥狀。其MRI表現與RDD相似,鑒別上有一定困難。
3.3.3 朗格漢斯組織細胞增生癥 又稱嗜酸性肉芽腫[14],好發于青少年,其所引起的骨質溶骨性改變常導致椎體變扁,多位于頸胸段,可形成軟組織腫塊,較少累及椎管內。T1WI呈等信號,T2WI呈稍高信號,增強掃描明顯強化。
3.3.4 脊柱結核 一般累及椎間隙為主的相鄰椎體及椎旁軟組織,邊界不清,椎體周圍可形成冷膿腫,T2WI 內部信號不均勻、邊緣呈等信號而內部膿腫呈高信號,形成干酪樣壞死時呈稍低信號,增強掃描強化不均勻,內部膿腫或者干酪樣壞死無強化。
3.3.5 非特異性炎性或肉芽腫性病變 如特發性肥厚性硬脊膜炎[15],是一種罕見的硬脊膜炎癥性疾病,其特征是硬脊膜發生肥厚性纖維增生,典型MRI 表現為沿硬脊膜生長的長條狀軟組織腫塊,T2WI序列上呈稍低信號,增強掃描邊緣呈均勻明顯強化,內部強化程度較輕。
3.4 治療和預后
主要治療手段包括手術切除、激素治療、化療和放射治療[16]。能否完整切除病灶對患者的預后至關重要,病灶刮除后需行病理學檢查明確診斷。本組4例病變局限于胸椎者行手術予以完全切除,隨訪期間復查MRI均未見復發跡象。
對無法完整切除病變的病例,無癥狀者可隨訪觀察,有癥狀者可聯合激素治療、放療或化療[17]。但放療劑量和周期仍未明確,激素及化療療效的可靠性和持久性也不確定[4,18]。本組對術后殘留病灶酌情給予輔助放療等治療,也有病例未做進一步治療,隨訪結果均滿意。
RDD 具有高度侵襲性,但也有學者認為其有一定的自限性,可自行緩解[19]。本組2例癥狀不明顯且位于寰樞椎者僅取部分病灶活檢,未做任何治療,隨訪期間復查CT及MRI提示骨質受侵情況較前好轉,骨質局部有硬化表現,臨床癥狀未見明顯加重,與國外自限性報道相一致[19]。
綜上,淋巴結外原發脊柱RDD 臨床罕見,有高度侵襲性,但亦有良性組織增生表現,具有一定的自限性;明確診斷需依靠病理學檢查,其影像學表現,特別是MRI 征象,可為該病的診斷、鑒別診斷、手術定位和預后提供重要的影像學依據。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
