吹掃捕集-氣相色譜質譜法測定水中反式-1,2-二氯乙烯的不確定度評定
2021-03-16 04:01:04李翠霞
中國新技術新產品 2021年24期
時 妍 李翠霞
(1.吉林市水務集團有限公司,吉林 吉林 132000;2.吉林市職業危害檢測檢驗中心,吉林 吉林 132000)
不確定度是根據所用到的信息來表征賦予被測量量值分散性的非負參數。通過檢測分析方法測量不確定度各分量的來源和大小,以評估檢測結果的準確性[1]。隨著工業的發展,水中污染物的種類,尤其是有機污染物也日益增多。1,2-二氯乙烯有2 個同分異構體,分別為反式-1,2-二氯乙烯和順式-1,2-二氯乙烯。其中,水中反式-1,2-二氯乙烯主要來自于工業污染,對人體有害[2]。因此,為確保水樣檢測結果的準確性和可靠性,測量不確定度的評定就顯得尤為重要。
目前,檢測有機物的方法有很多,其中,吹掃捕集儀簡化了樣品的前處理過程,使樣品分析更加簡便,氣相色譜質譜聯用儀(GC-MS)適合分析易揮發性、分子小、熱穩定且能氣化的化合物,而且所需試樣量少,分離效率高,靈敏度高,具有高選擇性的特點,應用范圍很廣,是唯一可以確定分子式的方法。將吹掃捕集儀和氣相色譜質譜聯用儀有機結合,可使其檢驗檢測優點最大化。因此,該文采用吹掃捕集-氣相色譜質譜法對反式-1,2-二氯乙烯進行檢測分析,然后根據《測量不確定度評定與表示》、《化學分析測量不確定度評定》及文獻[3]對吹掃捕集-氣相色譜質譜法測定水中反式-1,2-二氯乙烯的測量不確定度進行評定,明確了各不確定度分量對檢測結果的影響程度,為生活飲用水中揮發性有機物的含量檢測方法提供了技術支持。
1 檢測方法
1.1 原理
吹掃捕集儀的吹掃管通常用氦氣對水樣進行吹掃,將反式-1,2-二氯乙烯從水樣中吹脫出來,然后被裝有吸附劑的捕集阱捕獲,通過瞬間加熱捕集阱并用氦氣反吹,將所吸附的反式-1,2-二氯乙烯解吸入氣相色譜質譜聯用儀中進行分離測定。根據保留時間和計算機譜庫中的質譜圖進行定性,再通過定量離子強度和外標法繪制的標準曲線來定量。
1.2 主要儀器和試劑
試驗過程中相關儀器和試劑如下:1) 吹掃捕集為Tekmar 9800。2) 氣相色譜質譜聯用儀為安捷倫7890A-5975C。3) 吹掃捕集進樣器容量為5 mL。4) 微量進樣器容量為5 μL、25 μL、50 μL、100 μL 以及500 μL。5) 容量瓶容量為100 mL。6) 反式-1,2-二氯乙烯標準溶液購自中國計量科學研究院,溶劑為甲醇,標準值為0.881 mg/mL。7) 純水為超純水,空白樣品檢測無響應。8) 載氣為高純氦氣(99.999%以上)。
1.3 儀器參數條件
1.3.1 吹掃捕集條件
吹掃溫度為20 ℃,吹掃時間為5 min,解吸溫度為170 ℃,解吸時間為5 min,烘烤溫度為200 ℃,烘烤時間為5 min,吹掃流量為(40±5)mL/min。
1.3.2 氣相色譜質譜聯用儀條件
氣相色譜儀為DB-5MS 毛細管色譜柱。色譜柱升溫程序的起始溫度為35 ℃(保持2 min),以10 ℃/min 的速度升溫到55 ℃(保持1.5 min),再以30 ℃/min 的速度升溫到150 ℃(保持0 min)。柱流量為0.48 mL/min。分流比為20 ∶1;進樣口溫度為230 ℃。在質譜儀中,離子源(EI)溫度為230 ℃,四極桿溫度為150 ℃,接口溫度為250 ℃,離子化能量為70 eV,掃描范圍為35 u~300 u(u 為氣質聯用儀掃描單位),掃描時間為0.45 s,回掃時間為0.05 s,溶劑延遲時間為2 min。
1.4 水樣采集與保存
準備容量為100 mL 的棕色磨口采樣瓶,用超純水清洗采樣瓶數次。出廠水的采集需要打開水龍頭放水約10 min,達到水溫恒定,調節水流的流速為500 mL/min,向采樣瓶中以每40 mL 水樣加入25 mg 抗壞血酸穩定劑的量來加入相應的穩定劑,將水樣充滿采樣瓶至溢流,旋緊瓶蓋;水源水的采集需要用源水沖洗已洗凈的采樣瓶(數次),將水樣充滿采樣瓶至溢流,旋緊瓶蓋。將裝有水樣的采集瓶放置在4 ℃冰箱低溫保存,水樣存放區域要保證沒有有機物的干擾,在采集后14 d 內對水樣進行檢測分析。
1.5 操作步驟
根據《生活飲用水標準檢驗方法》(GB/T 5750.8—2006)有機物綜合指標附錄A 吹掃捕集/氣相色譜質譜法對反式-1,2-二氯乙烯進行測定。按照實驗室要求,儀器開機預熱至少4 h,以達到完全真空的狀態,然后進行GC-MS 性能測試,調諧并分析調諧報告,以滿足測定條件,否則重新調諧質譜儀,直到符合要求方可對樣品進行分析測定。對高濃度標準儲備液進行一級稀釋,配制成標準中間液。吸取100 μL 含有(反式-1,2-二氯乙烯)標準物質的儲備液加入100 mL 容量瓶中,然后用超純水定容,混勻后分裝到20 mL 頂空瓶中,注意瓶頂上端不留空間和氣泡,封蓋后可放置4 ℃冰箱中低溫保存。在試驗測定前,將裝有標準中間液的頂空瓶取出,與室溫平衡。分別吸取2.5 μL、5.0 μL、20.0 μL、50.0 μL、100.0 μL、250.0 μL 以及500.0 μL 的標準中間液,直接將其注入定容到裝有不含有機物超純水的5 mL 吹掃捕集進樣器中,混合均勻后進樣測定,即可配制成含有7 個標準濃度點的標準溶液。繪制標準曲線時,標準溶液要現用現配,要使該標準物質的國家標準限值高于標準曲線。每次只配制1 個標準溶液濃度點進行進樣分析測定,得到各標準溶液濃度值相對應的響應值,將所得各標準濃度點的數值進行數據處理,用最小二乘法進行擬合,最終得到回歸方程和相關系數。
2 數學模型的建立
標準曲線的回歸方程如公式(1)所示。
Y=aC+b(1)式中:Y為響應值;C為標準樣品的濃度;a為校準曲線的斜率;b為校準曲線的截距。
3 不確定度來源分析
根據整個試驗的操作過程可以得出以下4 個方面為測定反式-1,2-二氯乙烯過程中所引入的不確定度的主要來源:1) 配制標準曲線的過程所引入的不確定度urel(C1)。2) 最小二乘法擬合標準曲線的過程所引入的不確定度urel(C2)。3) 重復測量的過程所引入的不確定度urel(C3)。4) 加標回收率的過程中引入的不確定度urel(Rec)。
4 不確定度評定計算依據
該文主要根據《測量不確定度評定與表示》和《化學分析測量不確定度評定》中的方法對整個試驗過程中所引入的各不確定度來源進行分析計算。
5 不確定度的評定
5.1 配制標準曲線的過程中引入的不確定度urel(C1)
5.1.1 標準溶液自身所引入的不確定度u(C儲)
實驗室所用標準溶液是購自有標準物質生產資質的生產廠家,在該文所用反式-1,2-二氯乙烯標準儲備液的證書中,已給出相對擴展不確定度,即3%(k=2,k為包括因子),根據不確定度評定計算依據可知,標準溶液自身所引入的相對標準不確定度u(C儲)=0.03/2=0.015。
5.1.2 配制標準溶液系列過程所引入的不確定度u(V標)

在下文的相關公式中,ΔV為體積變化允許誤差;V為器具體積;ΔT為溫度變化允許誤差。
5.1.2.1 5 μL 微量進樣器所引入的不確定度urel(V5)
5.1.2.1.1 體積引入的不確定度u1(V5)

5.1.2.2 100 mL 容量瓶引入的不確定度urel(V容)
5.1.2.2.1 體積引入的不確定度u2(V容)

根據上述方法可以計算出該實驗中各微量進樣器、吹掃捕集進樣器和容量瓶所引起的標準不確定度和相對標準不確定度,根據繪制標準曲線配制各濃度點的實際過程可知各器具所使用的次數,見表1。
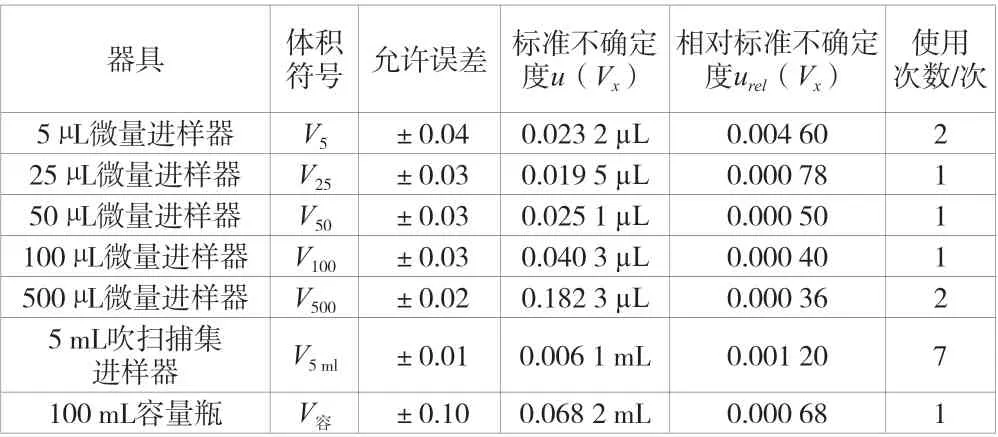
表1 該實驗中進樣器和容量瓶所引起的標準不確定度和相對標準不確定度
根據上述數據和不確定度評定計算依據可以計算出配制標準溶液系列產生的相對標準不確定度urel(C標)為各項相對標準不確定度的平方與使用次數相乘的和再開平方,計算得urel(C標)=0.007 4。因此,配制標準曲線過程所引入的不確定度urel(C1)為u(C儲)和urel(C標)的平方和,再開平方得出urel(C1)=0.016 7。
5.2 最小二乘法擬合標準曲線的過程所引入的不確定度urel(C2)
建立了7 個濃度點的標準曲線,結果見表2。分別取50 μL標準中間液注入裝有不含有機物超純水的5 mL 吹掃捕集進樣器中,配置成一定濃度的待測樣品并進行7 次獨立的重復測定,計算得相對標準偏差,結果見表3。
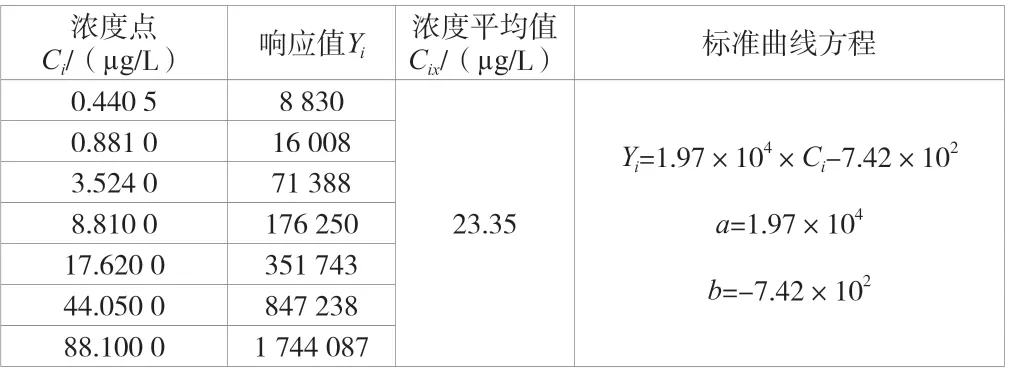
表2 標準曲線各濃度點下的響應值
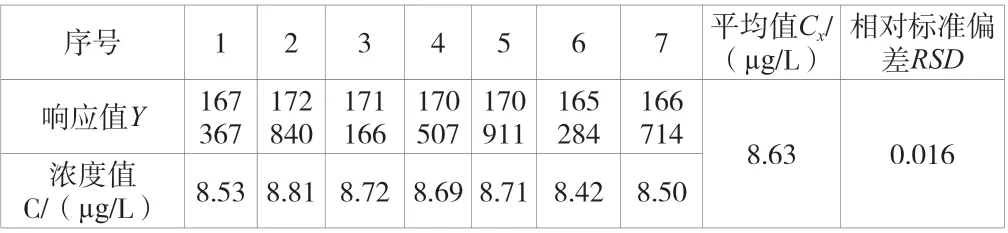
表3 樣品測定結果
根據不確定度評定計算依據可查得公式,將標準曲線各值代入公式(1),計算得標準不確定度u(C2)=0.295 8 μg/L。式中:SR為回歸曲線剩余標準差(殘差的標準差)。
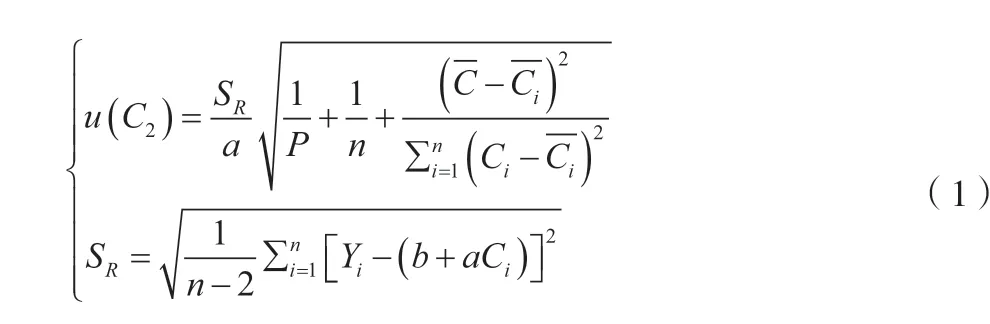
因此,最小二乘法擬合標準曲線的過程所引入的不確定度urel(C2)=0.2958/8.63 ≈0.034 3。
5.3 重復測量的過程所引入的不確定度urel(C3)
對待測樣品濃度進行7 次獨立重復測定,測定的平均值為Cx=8.63 μg/L,結果為A 類評定,其結果見表3。根據不確定度評定計算依據,計算得標準不確定度u(C3)=0.1423 μg/L。因此,重復測量的過程所引入的不確定度urel(C3)=0.1423/8.63 ≈0.016 5。
5.4 加標回收率的過程所引入的不確定度urel(Rec)
加標回收率引入的不確定度主要來源為吹掃捕集前處理吹掃、轉移進入GC-MS 的過程中。向空白水樣中分別加入20 μL、50 μL 以及250 μL 的標準中間液,配置成分別含有3.524 μg/L、8.81 μg/L 以及44.05 μg/L 濃度的3 個加標樣品,對3 個加標樣品進行加標回收率試驗并計算得各加標濃度下的加標回收率,測定結果見表4。
由表4 可知,反式-1,2-二氯乙烯的加標回收率為88.7%~95.3%,平均回收率為92.5%。因此,根據不確定度評定計算依據,加標回收率的過程所引入的不確定度urel(Rec)=0.019 1。
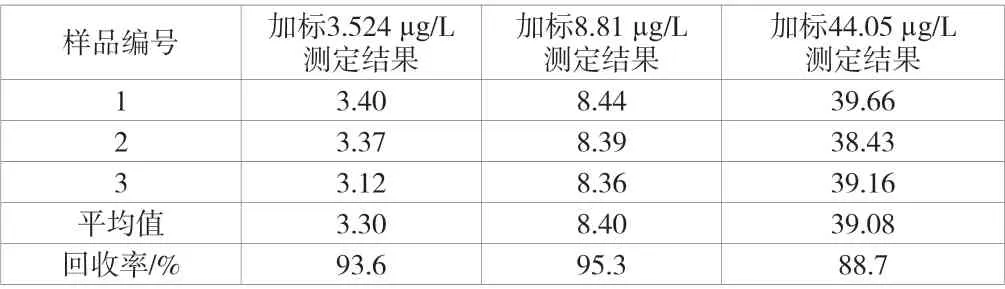
表4 樣品加標回收實驗測定結果
6 合成標準不確定度U(C)
將所引入的4 個方面的不確定度來源的計算結果匯總,見表5。

表5 相對標準不確定度計算結果匯總
根據檢測方法,各不確定度分量之間相互獨立,參照不確定度評定計算依據,計算各相對標準不確定度的平方和再開平方得到urel(C)=0.045 7。因此,其合成相對標準不確定度U(C)=urel(C)×Cx=0.0457×8.63 ≈0.394 4 μg/L。
7 擴展不確定度U
一般取包括因子k=2(置信概率為95%),參照不確定度評定計算依據可知,擴展不確定U=U(C)×k,則U=0.3944×2=0.79 μg/L。
8 最后測量結果報告
水中反式-1,2-二氯乙烯含量的測量結果如下。C=(8.63±0.79) μg/L (k=2)
9 結語
該文對吹掃捕集-氣相色譜質譜法測定水中反式-1,2-二氯乙烯的測量不確定度評定進行研究。通過表5 可知,對不確定度影響來源由大到小依次為最小二乘法擬合標準曲線、加標回收率、配置標準曲線以及重復測量。其中,最小二乘法擬合標準曲線對不確定度的影響最大,因此,在實驗中應注意微量進樣器在吸取標準中間液及定容至5 mL吹掃捕集進樣器的操作過程,要提高操作的規范性,降低不確定度。同時,該不確定度評定的方法可以為在同方法、同條件水樣中其他揮發性有機物的檢測提供測量結果不確定度的計算依據,確保實驗室分析測試的數據準確性、可靠性。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·八年級物理人教版(2019年9期)2019-11-25 07:33:02
當代陜西(2019年8期)2019-05-09 02:22:48
中學生數理化·八年級物理人教版(2019年3期)2019-04-25 06:20:54
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
中學生數理化·八年級物理人教版(2018年3期)2018-05-31 08:52:45
海峽科技與產業(2016年3期)2016-05-17 04:32:12
