UPLC測定經典名方金水六君煎中11種成分
2021-02-03 07:45:08董自亮李紅亮原歡歡蔣燕霞禹奇男吳瑞軍冉亞東安太勇彭世陸劉世琪秦少容羅維早
中草藥 2021年3期
關鍵詞:質量
董自亮,李紅亮,原歡歡,蔣燕霞,李 娟,禹奇男,吳瑞軍,冉亞東,安太勇,覃 瑤,彭世陸,劉世琪,彭 濤,秦少容,羅維早*,王 欣*
1.重慶市中藥研究院,重慶 400065
2.太極集團有限公司,重慶 400800
金水六君煎(Jinshui Liujun Decoction,JLD)出自明·張介賓《景岳全書》,為國家中醫藥管理局2018年公布的《古代經典名方目錄(第一批)》的經典名方的第58 首。原文:“當歸二錢,熟地三五錢,陳皮一錢半,半夏二錢,茯苓二錢,炙甘草一錢,水二鐘,生姜三五七片,煎七八分,食遠溫服”。方中半夏辛溫,能燥濕化痰、和中止嘔;陳皮芳香,理氣運脾、燥濕化痰;茯苓甘淡,甘能補脾,淡可滲濕,使已聚之濕從小便滲利而去,更添甘草和中益脾,共奏理氣健脾、燥濕化痰之效。主治肺腎虛寒、水泛為痰、或年邁陰虛、血氣不足、外受風寒、咳嗽嘔惡、喘逆多痰等癥[1]。近年來對該方的藥效學研究較多,但對其化學成分的研究及定量測定報道較少。文獻僅報道采用HPLC 對JLD 制劑中陳皮(橙皮苷)、當歸(阿魏酸)藥材進行定量測定[2-3],以及采用薄層掃描法、紫外分光光度法測定JLD 膠囊中β-谷甾醇和總生物堿的含量[4],但未能全面有效地控制該復方制劑的多種成分。本研究采用UPLC 技術[5-6],首次建立了多波長同時測定JLD 中腺苷、鳥苷、5-羥甲基糠醛、阿魏酸、甘草苷、毛蕊花糖苷、蕓香柚皮苷、橙皮苷、甘草酸銨、6-姜辣素、藁本內酯11 種主要成分的UPLC 方法,此方法簡便、快捷、準確,專屬性強,同時結合偏最小二乘法-判別分析(PLS-DA)等化學識別模式[7],評價各批次樣品之間的質量差異,在此基礎上為Hotelling’sT2和DModX 2 種統計量設定了控制限和警戒限,為JLD 物質基準的關鍵質量屬性的研究及其后續現代制劑質量的全面控制提供參考。
1 儀器與材料
1.1 儀器
Agilent 1290-UPLC DAD 檢測器,美國Agilent公司;XSE205DU 型電子天平,精密度0.01 mg/0.1 mg,Mettler Toled 公司;LGJ-10FD 型冷凍干燥機,北京松源華興科技發展有限公司;KQ-500DE 超聲波儀,昆山市超聲儀器有限公司。
1.2 材料
JLD,實驗室自制;對照品腺苷(批號110879-201703,質量分數99.7%)、鳥苷(批號111977-201501,質量分數93.6%)、5-羥甲基糠醛(批號111626-201912,質量分數99.2%)、阿魏酸(批號110773-201915,質量分數99.4%)、甘草苷(批號111610-201908,質量分數95.0%)、毛蕊花糖苷(批號111530-201914,質量分數95.2%)、橙皮苷(批號110721-201818,質量分數96.2%)、甘草酸銨(批號110731-201720,質量分數97.7%)、6-姜辣素(批號111833-201806,質量分數99.9%)、藁本內酯(批號111737-201910,質量分數≥98%),均購于中國食品藥品檢定研究院;對照品蕓香柚皮苷,批號18040301,質量分數99.66%,購自上海詩丹德標準技術服務有限公司。乙腈、磷酸均為色譜純,北京化標源科技有限公司;實驗用水均為二次蒸餾水。
JLD 中各飲片均采自3 個產地,按《中國藥典》2015年版一部項下檢測均合格。產地、批次等具體信息見表1。經重慶市中藥研究院羅維早研究員鑒定,各飲片均符合《中國藥典》2015年版一部相關項下的性狀規定,鑒定結果地黃為玄參科地黃屬植物地黃Rehmannia glutinosaLibosch 的新鮮或干燥塊根,當歸為傘形科當歸屬植物當歸Angelica sinensis(Oliv.) Diels 的干燥根,陳皮為蕓香科柑橘屬植物橘Citrus reticulataBlanco 的干燥成熟果皮,半夏為天南星科半夏屬植物半夏Pinellia ternata(Thunb.) Breit.的干燥塊莖,茯苓為多孔菌科茯苓屬真菌茯苓Poria cocos(Schw.) Wolf 的干燥菌核,豆科甘草屬植物甘草Glycyrrhiza uralensisFisch.的干燥根和根莖,姜科姜屬植物姜Zingiber officinaleRose.的新鮮根莖。
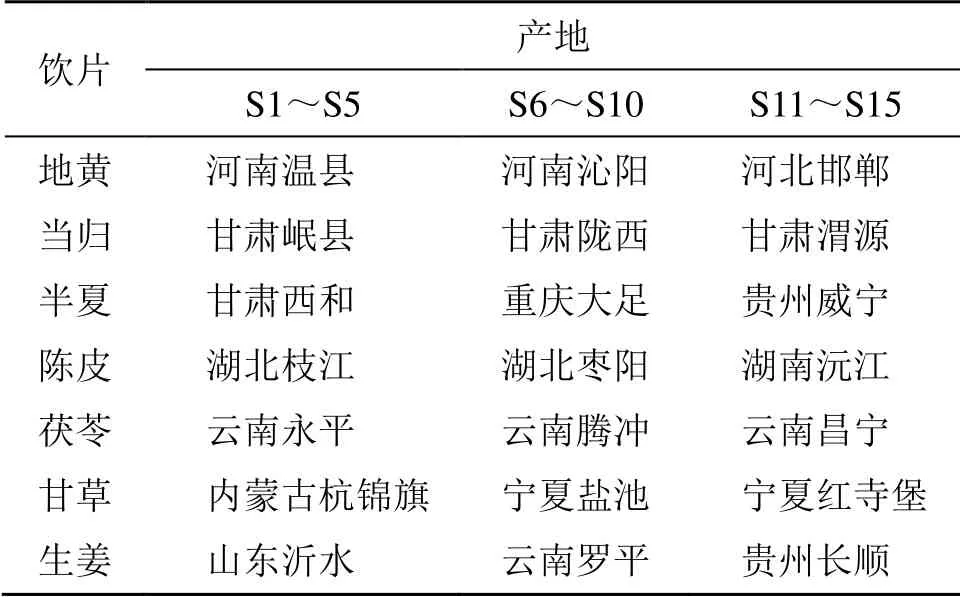
表1 15 批次JLD 樣品各批次中所用飲片的產地信息Table 1 Origin information of decoction pieces used in each batch of 15 batches of JLD samples
2 方法與結果
2.1 色譜條件與系統適應性
色譜柱為Acquity UPLC HSS T3 C18柱(100 mm×2.1 mm,1.8 μm);流動相為0.1%磷酸水溶液(A)-乙腈(B),梯度洗脫:0~2 min,100% A;2~7 min,100%~98% A;7~12 min,98% A;12~25 min,98%~90.5% A;25~32 min,90.5%~85.5% A;32~41 min,85.5% A;41~50 min,85.5%~79% A;50~58 min,79%~71% A;58~64 min,71%~44%A;64~70 min,44%~35% A;70~71 min,35%~5% A;72 min,5% A;72~73 min,5%~100% A;體積流量0.3 mL/min;檢測波長:248 nm(0~17 min、64~66 min)、330 nm(18~40 min)、281 nm(40~64 min、66~73 min)切換;柱溫25 ℃;進樣量1.0 μL。各峰分離度均大于1.5。色譜圖見圖1。
2.2 對照品溶液的制備

圖1 空白溶劑 (A)、混合對照品溶液 (B)、供試品溶液 (C)UPLC 圖Fig.1 UPLC of blank (A),standard solution (B) and sample solution (C)
精密稱取腺苷、鳥苷、5-羥甲基糠醛、阿魏酸、甘草苷、毛蕊花糖苷、蕓香柚皮苷、橙皮苷、甘草酸銨、6-姜辣素、藁本內酯對照品適量,加甲醇溶解后配制成質量濃度分別為40.12、69.16、738.80、39.88、193.80、58.75、164.60、421.80、218.90、134.90、25.20 μg/mL 的混合對照品儲備液,保存于4 ℃冰箱中備用。
2.3 供試品溶液的制備
采用隨機數表法,分別對熟地黃、當歸、陳皮、半夏、茯苓、炙甘草、生姜各15 批飲片進行編號及排序。根據原方記載及本方考證的劑量折算關系,稱取一個處方量的JLD(當歸7.46 g、熟地黃14.92 g、陳皮5.60 g、半夏7.46 g、茯苓7.46 g、炙甘草3.73 g、生姜15 g),加水400 mL,加熱至沸騰轉小火,保持沸騰30~60 min,濾過,濾液冷凍干燥(-50 ℃干燥72 h,5 Pa),即得JLD 干粉。
取本品約0.5 g,加70%甲醇15 mL,置于密塞錐形瓶中,精密稱定質量,超聲提取45 min,放冷,用70%甲醇補足減失的質量,搖勻,濾過,即得供試品溶液。
2.4 方法學考察
2.4.1 線性關系考察 取“2.2”項下混合對照品儲備液,分別精密吸取1、2、3、5、10、20 mL 置于25 mL 量瓶中,加甲醇至刻度,搖勻,即得不同質量濃度的混合對照品溶液;按“2.1”項色譜條件進樣測定。以對照品的質量為橫坐標(X),峰面積積分值為縱坐標(Y),繪制標準曲線,進行線性回歸,得到回歸方程和相關系數(r),結果見表2。
2.4.2 精密度試驗 取“2.4.1”項下混合對照品溶液,按“2.1”項色譜條件連續進樣6 次,結果腺苷、鳥苷、5-羥甲基糠醛、阿魏酸、甘草苷、毛蕊花糖苷、蕓香柚皮苷、橙皮苷、甘草酸銨、6-姜辣素、藁本內酯峰面積的RSD 值分別為1.26%、1.45%、1.06%、0.17%、0.95%、1.43%、0.86%、1.24%、0.23%、1.26%、0.95%。11 種成分峰面積的RSD 值均小于2.0%,表明儀器精密度良好。
2.4.3 穩定性試驗 取1 份批號為S1 的樣品,按“2.3”項下方法制備供試品溶液,分別于第0、6、12、18、24 h 按“2.1”項下色譜條件進行測定。結果腺苷、鳥苷、5-羥甲基糠醛、阿魏酸、甘草苷、毛蕊花糖苷、蕓香柚皮苷、橙皮苷、甘草酸銨、6-姜辣素、藁本內酯峰面積的RSD 值分別為1.92%、2.68%、0.98%、1.18%、0.90%、1.23%、0.51%、1.43%、0.85%、1.55%、0.95%,結果發現11 種成分峰面積的RSD 值均小于3.0%,表明JLD 供試品溶液在24 h 內穩定性良好。

表2 11 種成分的線性回歸方程、線性范圍、相關系數Table 2 Regression equation,linear range and correlation coefficient of 11 active ingredients
2.4.4 重復性試驗 取JLD 樣品(批號為S1),按“2.3”項下方法制備6 份供試品溶液,按“2.1”項下色譜條件進樣測定。結果腺苷、鳥苷、5-羥甲基糠醛、阿魏酸、甘草苷、毛蕊花糖苷、蕓香柚皮苷、橙皮苷、甘草酸銨、6-姜辣素、藁本內酯質量分數的RSD 值分別為2.88%、1.69%、2.35%、0.95%、0.53%、1.05%、0.56%、1.85%、0.40%、0.36%、1.81%,11 種成分質量分數的RSD 值均小于3.0%,表明該分析方法重復性良好。
2.4.5 加樣回收率試驗 取已測定含量的JLD樣品(批號為S1),稱取約0.25 g,精密稱取6 份,分別加入約為樣品中等量的各對照品,按“2.3”項下方法制備供試品溶液,按“2.1”項下色譜條件進樣測定,記錄峰面積并計算各成分質量濃度,計算加樣回收率。結果腺苷、鳥苷、5-羥甲基糠醛、阿魏酸、甘草苷、毛蕊花糖苷、蕓香柚皮苷、橙皮苷、甘草酸銨、6-姜辣素、藁本內酯的平均加樣回收率分別97.70%、103.15%、100.71%、97.79%、99.74%、102.72%、96.62%、101.43%、102.35%、97.65%、98.11%,RSD 值分別為0.78%、1.67%、1.04%、0.50%、1.60%、0.68%、0.98%、0.56%、0.19%、1.54%、0.23%。
2.5 樣品測定
按“2.3”項下樣品處理方法,制備15 批供試品溶液,按照“2.1”項下色譜條件進行測定,采用外標法計算JLD 中腺苷、鳥苷、5-羥甲基糠醛、阿魏酸、甘草苷、毛蕊花糖苷、蕓香柚皮苷、橙皮苷、甘草酸銨、6-姜辣素、藁本內酯的質量分數,結果見表3。

表3 15 批JLD 樣品中11 種成分測定結果Table 3 Content of 11 components in 15 batches of JLD
2.6 PLS-DA 模型的建立及相關分析
采用 SIMCA 14.1 軟件(Umetrics,MKS Instruments Inc.,Sweden)對15 批JLD 樣品中的多指標含量測定數據進行主成分分析(PCA)。應用Hotelling’sT2和DModX 2 種控制圖對不同批次的樣品質量進行監測,進而采用有監督的PLS-DA 模型進行分析。
2.6.1 Hotelling’sT2和 DModX 控制限的建立Hotelling’sT2和DModX 控制圖是為了監測不同批次產品而開發的,是2 個互補的多變量分析手段。Hotelling’sT2表示的是每個選定觀察點與模型平面中原點的距離,為模型的內部變化度量,代表樣品中信息與主成分模型中其他樣品差異;DModX 表示數據在變量X空間到主成分模型的距離,為模型外部的數據變化度量,代表樣品中未被模型解釋的變化。通常認為在控制限度以下的產品為正常批次產品,超出控制限的為異常批次樣品。Hotelling’sT2和DModX 統計量作為批間差異性評價指標,兩者具有不同的監控作用,互為補充。當發現某個批次超出或控制限時,在貢獻圖上可以分析批次過程的異常波動受哪些變量的影響[8]。
Hotelling’sT2和DModX 的控制圖如圖2、3,圖中的T2Crit (99%)和DCrit 為控制限,其控制上限分別為14.43 和1.75;T2Crit (95%)為警戒限,上限為8.20。這15 批物質基準樣品在控制限之下,均為正常批次產品,在2 張控制圖上處于受控狀態,這也證明所建立的PCA 模型較好的表征了樣品的正常波動。說明這15 批物質基準樣品批次質量差異性較好,與相似度評價結果相對應,并且較于相似度閾值解釋性更強。
2.6.2 偏最小二乘判別分析(PLS-DA) 為評價不同批次JLD 樣品之間的成分含量差異,采用有監督的PLS-DA 模型進行分析[9]。以樣品S1~S15 為Y變量,11種成分的含量為X變量,利用SIMCA 14.1軟件進行PLS-DA 處理,見圖4、5。結果發現在置信區間(95%)內,15 批樣品存在一定差異性,根據分布可將樣品S1~S15 分為3 類,樣品S1~S5為一類,S6~S10 為一類,S11~S15 為一類,表明這11 種成分在評價樣品產地之間的質量差異具有重要參考價值。同時,變量重要性投影(VIP)值越大,表明對樣品分類貢獻較大;其中VIP>1 的有4 種成分,依次為蕓香柚皮苷、甘草酸銨、藁本內酯和阿魏酸,說明這4 種成分是影響不同批次樣品之間質量差異貢獻較大的成分,可作為指導經典名方JLD 物質基準及復方制劑質量控制的關鍵成分。

圖2 15 批JLD 樣品Hotelling’s T2 控制圖Fig.2 Hotelling’s T2 control chart of 15 batches of JLD

圖3 15 批JLD 樣品的DModX 控制圖Fig.3 DModX control chart of 15 batches of JLD

圖4 15 批JLD PLS-DA 的得分Fig.4 PLS-DA score plot of 15 batches of JLD

圖5 JLD 中11 種成分的VIP 值Fig.5 VIP value of 11 components of JLD
3 討論
3.1 指標成分的確定
本研究在《中國藥典》對JLD 各藥味明確規定的甘草苷、甘草酸、毛蕊花糖苷、橙皮苷、阿魏酸、6-姜辣素等6 個定量指標的基礎上增加了與該方藥效相關的腺苷、鳥苷、5-羥甲基糠醛、蕓香柚皮苷、藁本內酯5 個關鍵成分的定量控制。地黃為本方君藥,其中毛蕊花糖苷、5-羥甲基糠醛等活性成分具有免疫調節、肝臟保護以及神經保護等作用[10]。陳皮的主要活性成分是橙皮苷、蕓香柚皮苷、柚皮苷,具有較好的抗氧化能力、抗炎、促胃動力作用、抗腫瘤活性[11-14]。當歸中阿魏酸、藁本內酯等活性成分具有抗炎止痛、抗血小板凝聚等活性[15];甘草中的甘草酸、甘草黃酮為生物活性較強的物質,現代藥理學研究表明甘草酸具有較強的保肝活性和腎上腺皮質激素樣作用,可增強內耳聽覺,能夠抗炎、抗變態反應、解毒止咳、抗腫瘤等作用。甘草中的甘草苷具有抗潰瘍、抗菌、抗炎、解痙、鎮痛等作用[16-20]。尿苷、鳥苷、腺苷等核苷類成分能夠干擾病毒核酸的合成,通常為中成藥抗病毒的活性成分[21]。綜合藥理作用與色譜分離效果,最終確定該11個成分為指標成分。
3.2 色譜條件的選擇
中藥復方化學成分較為復雜,選擇梯度洗脫能更有利于成分整體信息的獲得。考察供試品溶液制備時不同提取溶劑(甲醇、乙醇及水)、不同體積分數(30%、50%、70%、100%)甲醇和水的配比、不同提取時間(30、45、60 min)、提取溶劑體積(10、15、20、25 mL)對JLD 中有效成分的影響,綜合考慮各待測成分的色譜峰峰形及分離度,最終選擇70%甲醇為JLD 干粉的提取溶劑。
考察了乙腈和甲醇作為有機相對提取效果的影響,結果甲醇作有機相基線漂移,峰形不對稱,因此選擇乙腈作有機相;為改善峰形,在水相中加入不同濃度的甲酸、乙酸和磷酸,結果表明加入0.1%磷酸時,分離效果好,基線平穩;考察了梯度洗脫和等度洗脫,梯度洗脫時各物質分離度高。
根據對照品預試驗初步考察,腺苷、鳥苷、5-羥甲基糠醛、阿魏酸、甘草苷、毛蕊花糖苷、蕓香柚皮苷、橙皮苷、甘草酸銨、6-姜辣素、藁本內酯的最佳吸收波長分別為260、256、285、322、276、330、283、283、252、280、281 nm,由于本研究測定成分較多,成分間波長差異較大,為實現多成分含量同時測定,在綜合考察峰形、基線、吸收差異等多項干擾因素下,最終確定最佳的波長切換法對多成分進行含量測定。
3.3 結果分析
本研究在UPLC 多成分含量測定基礎上,結合PCA、PLS-DA 等相關化學計量學方法對不同批次JLD 物質基準的質量的批間差異性進行了深入的分析,并在PCA 模型基礎上設定了Hotelling’sT2和DModX 控制限,實現對批次物質基準質量進行監測。該15 批物質基準均為正常批次樣品,其有效成分含量變化都沒有明顯的差異,但通過PCA 后可以看出,15 批樣品所用原藥材產地的差異在多指標成分的含量上予以體現;進一步通過PLS-DA中的VIP值可以判斷出,蕓香柚皮苷(源于陳皮)、甘草酸銨(源于甘草)、藁本內酯(源于當歸)和阿魏酸(源于當歸、陳皮)是造成這些差異存在的主要原因,提示對這4 個成分進行質量控制,有利于有效反映JLD 物質基準及其復方制劑的質量變化屬性。
最后,本研究以JLD 中腺苷、鳥苷、5-羥甲基糠醛、阿魏酸、甘草苷、毛蕊花糖苷、蕓香柚皮苷、橙皮苷、甘草酸銨、6-姜辣素、藁本內酯等11 種主要成分為研究對象,首次建立了UPLC 同時測定JLD 中11 種成分的方法,同時對其提取條件、色譜條件等參數進行了優化,并通過對15 批不同產地的藥材進行對比分析,發現了影響JLD 物質基準質量較大的4 種成分。
方法學考察結果表明,所建立的方法滿足定量要求,且簡單易行,為JLD 的多指標質量控制提供了方法基礎,同時也為后續的制劑、藥效學研究提供了參考。
利益沖突所有作者均聲明不存在利益沖突
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
