抗富亮氨酸膠質瘤失活蛋白1抗體相關的神經性肌強直1例并文獻復習
2020-12-21 01:44:46解學軍劉艷麗李文靜喬曉東張志華李淑君趙偉麗
中風與神經疾病雜志 2020年10期
關鍵詞:血清
解學軍,劉艷麗,李文靜,喬曉東,張志華,李淑君,趙偉麗
本文報道1例臨床表現為肌肉顫搐、血清LGI1抗體陽性,診斷為抗LGI1抗體相關的神經性肌強直的病例,并通過復習文獻論述其病因及發病機制。
1 臨床資料
患者,女性,30歲,慢性病程,進行性加重;主因“發現肌肉蠕動1 y余”入院;1 y前無明顯誘因出現雙下肢乏力、酸痛,小腿肌肉蠕動,發作性、靜止狀態出現,疲勞后明顯,睡眠時有發作并伴有肌肉痙攣,未在意。11 m前“感冒”后雙側小腿肌肉蠕動發作頻繁,持續時間明顯延長(每次發作時間大于2 min),左側較右側明顯,逐漸出現雙側大腿、上肢近端、肩背部偶發肌肉蠕動,但程度輕、持續時間短(發作時間小于10 s),不伴有無力、麻木。病程中無心慌、手抖;無出汗、體重減輕,飲食睡眠好。既往甲狀腺功能減退,口服優甲樂25 μg 日一次,已停用6 m。無家族史。入院查體:Bp130/73 mmHg,心率65次/min,內科查體無異常。雙側腓腸肌肉眼可見蠕動,叩擊無肌丘。神經系統查體:神清語利,高級智能正常,顱神經未見異常,四肢肌力5級,腱反射對稱引出,深淺感覺正常,病理反射未引出。輔助檢查:血、尿便常規、肝腎功能均正常;糖化血紅蛋白正常;肝炎分型(-)、HIV(-)、TPAb(-);血沉、CRP、PCT指標均正常;免疫全套篩查為陰性;女性腫瘤標記物9項篩查均正常范圍;葉酸、維生素B12正常;免疫固定電泳未見M蛋白。肌酶正常。甲狀腺功能:抗甲狀腺過氧化物酶抗體(anti-TPO Ab)136.800 IU/ml(0~34)升高、抗甲狀腺球蛋白抗體(anti-TG Ab)大于4000.0 IU/ml(0~115)明顯升高,促甲狀腺激素(TSH)10.350 mIU/L(健康人群0.27~4.2),T3、FT3、T4、FT4均正常;腦脊液:壓力120 mmH2O,無色清亮,細胞總數為2×106/L,腦脊液GLU2.48 mmol/L(2.5~4.5),腦脊液總蛋白315 mg/L(200~400),腦脊液CL 125.1 mmol/L(120~130)。同步血糖4.26 mmol/L(3.9~6.1),血CL106 mmol/L(99~110)。血清(serum)LGI1-Ab(+)(1∶32)、Caspr2-Ab(-);腦脊液(CSF)LGI1-Ab(-)、Caspr2-Ab(-)(北京協和醫院)。甲狀腺超聲:甲狀腺形態、大小正常,實質回聲增粗、減低、欠均勻,CDFI:血流分布及頻譜未見異常。乳腺超聲、腹部、泌尿系、婦科超聲均未見異常。胸部CT掃描未見異常。頭部MRI平掃及MRA未見明顯異常(見圖1)。腦電圖:清醒、睡眠視頻腦電圖正常腦電圖。肌電圖:神經傳導速度正常;RNS未見異常。針極電圖:插入所檢肌肉:雙側所檢肌肉靜息狀態下可見大量自發的呈二聯、三聯、多聯波。F波可見后放電現象(見圖2、圖3)。MoCA:24分。診斷:抗LGI1抗體相關的神經性肌強直。治療:靜脈注射用人丙種球蛋白: 0.4 g/(kg·d)×5 d,癥狀無明顯改善;醋酸潑尼松片:60 mg/d,每周減5 mg,減至10 mg持續口服1 m。口服潑尼松片3 d后發作頻率稍有減輕;卡馬西平片 100 mg bid 口服,3 d后無不適后加量至200 mg bid。隨訪:1 m后隨訪患者肌肉未再次出現蠕動;6 m后電話隨訪無發作;患者潑尼松片規律減停后同時停服卡馬西平片。
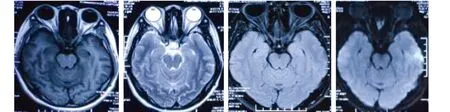
圖1 頭部磁共振成像(T1、T2、Flair DWI):未見異常
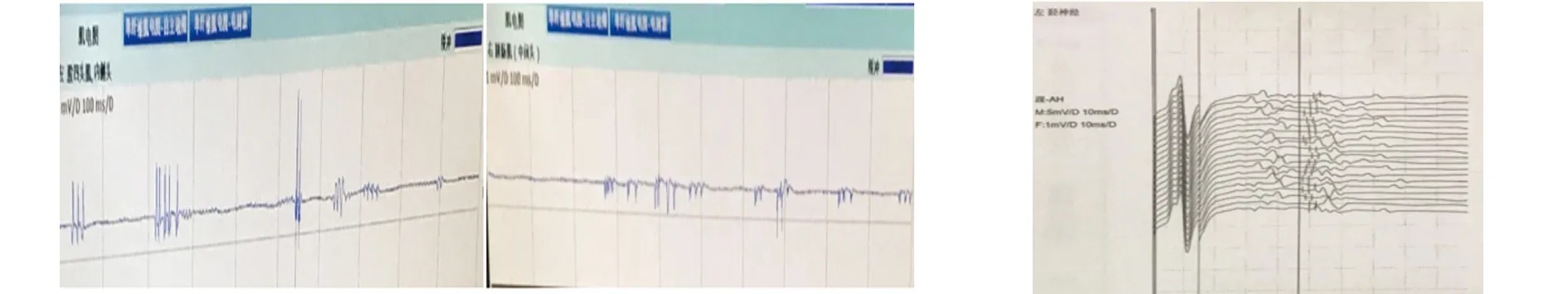
圖2 肌電圖:靜息狀態下可見大量自發的呈二聯、三聯、多聯波 圖3 F波后放電現象
2 討 論
本例患者因雙下肢肌肉顫搐(蠕動)就診,針極電圖:(1)肌肉靜息狀態下自發快速二聯、三聯、多聯的運動單位放電活動,肌顫搐電位(Myokymia);(2)F波后放電現象(after discharges)。符合神經性肌強直肌電圖表現。神經性肌強直(neuromyotonia,NMT)發病機制目前尚不完全清楚。Hart等[1]研究發現NMT患者體內可檢出一種或多種抗鉀離子通道抗體,這一結果不僅支持:NMT是一種免疫相關疾病,也支持它是一種免疫介導的離子通道病。Arimura等[2]指出,抗體所致的快速鉀離子外向電流的減少導致了周圍神經高興奮性。電壓門控鉀通道(voltage-gated potassium channels,VGKC)存在于中樞和外周神經系統的神經元膜上,在動作電位發生之后使細胞恢復到靜息狀態中發揮著至關重要的作用。致病抗體并非是針對VGKC,而是針對VGKC復合物相關蛋白:富亮氨酸膠質瘤失活蛋白1(1eucine-rich glioma-inactivated 1,LGI 1)和接觸蛋白相關蛋白2(contactin-associated Protein-like 2,Caspr2)。根據病因,NMT分為遺傳性NMT、獲得性NMT及特發性NMT 3類,NMT大多為獲得性。Caspr2抗體陽性的獲得性神經肌強直(Acquired neuromyotonia aNMT)、莫旺綜合征(Morvan’s syndrome)和痛性痙攣-束顫綜合征(cramp-fasciculation syndrome)患者具有周圍神經高興奮性(peripheral nerve hyperexcitability,PNH)[3];持續的后放電也作為周圍神經高興奮性的標志[4]。Irani等[5]研究證明aNMT患者CASPR2(contactin-associated protein-2)抗體陽性率40%,合并腫瘤者CASPR2抗體陽性率幾乎為100%。
但本例神經性肌強直患者,血清LGI1抗體(+),腦脊液LGI1抗體(-),血清與腦脊液CASPR2抗體(-)。LGI1抗體與神經性肌強直有關聯嗎?文獻檢索發現針對LGI1抗體與神經性肌強直描述或研究少之又少。Sneha I等[6]發現3例血清LGI1和 caspr2抗體陽性的神經性肌強直患者中除了1例伴有失眠外,未出現莫旺綜合征和lGI1抗體相關的中樞神經系統癥狀(如腦病、認知功能障礙、神經精神癥狀、邊緣性腦炎和癲癇)。但3例患者均為兒童(13歲,14歲,4歲),其臨床表現與美國梅奧診所發現的抗體雙陽性兒童病例表現相似[7]。Niu等[8]報告1例男性66歲患者,臨床表現為肌肉顫搐,無中樞神經系統癥狀;針極肌電圖提示肌顫搐電位、后放電現象,血清LGI1抗體陽性、腦脊液LGI1抗體陰性,并且血清、腦脊液CASPR2抗體均為陰性。Hor JY等[9]報告1例伴有胸腺瘤的重癥肌無力患者,免疫抑制劑治療(激素減量減至5 mg/d)過程中出現臨床癥狀表現認知功能障礙、人格改變,下肢肌肉顫搐、無力;針極肌電圖顯示二聯、三聯和多聯波;血清LGI1抗體陽性,CASPR2、NMDAR、AMPAR and GABA(B)R抗體均為陰性;診斷伴有肌強直(myokymia)的抗LGI1抗體相關腦炎。
上述病例報道肌肉顫搐均在免疫干預治療后得到緩解,說明神經細胞膜鉀離子通道障礙導致的周圍神經高興奮性機制中LGI1抗體也參與其中。但這需要我們更多的觀察和研究。獲得性NMT又可分為免疫相關性、腫瘤相關性、其他原因所致等。免疫相關性居多,可與多種自身免疫性疾病相共存[10],高達28%的成人LGI1和caspr2抗體陽性的患者中同時發現其他自身免疫性疾病。其發病機制尚不明確。國內有LGl1抗體相關的自身免疫性腦炎合并橋本腦病的文獻報道[11,12]。彭麗丹等學位論文病例資料中描述:3例抗LGI1抗體相關腦炎患者的甲狀腺自身抗體異常。并且石珂等[13]發現1例患者經激素治療后其甲狀腺抗體也基本恢復正常。本例患者抗甲狀腺過氧化物酶(anti-TPO)升高、抗甲狀腺球蛋白(anti-TG)明顯升高,甲狀腺功能正常,甲狀腺超聲實質回聲增粗、減低、欠均勻,支持橋本甲狀腺炎(HT)診斷。盡管本例患者未表現LGl1抗體相關的腦炎、橋本腦病典型臨床表現,但也提示自身免疫功能紊亂狀態下免疫抑制能力的減退。我們推斷:同一個體中多種自身免疫疾病共存,存在遺傳易感性;如人白細胞抗原(HLA)與HT和LGI1強相關性。Binks等[14]、van Sonderen等[15]研究發現LGI1和Caspr2抗體相關疾病與人白細胞抗原(human leucocyte antigen,HLA)強相關性;國內張琴等[16]研究發現自身免疫性甲狀腺疾病的患兒發病與HLA-DRB1、HLA-DQB1基因多態性明顯關聯。獲得性NMT另一病因及發病機制是腫瘤相關性所致等。神經系統副腫瘤綜合征(paraneoplastic neurological syndromes,PNS)是腫瘤極少見的遠隔效應所引起的,與腫瘤浸潤或轉移無關[17]。其臨床表現可以累及中樞神經系統、外周神經系統、神經肌肉接頭和肌肉的任何部位,這些部位可以是孤立的,也可以是疊加,大約25%的患者會有不同綜合征的重疊[18]。目前LGI-1抗體腦炎與腫瘤的關系尚不清楚,大多數LGI-1抗體腦炎患者不伴發腫瘤。本患者行腫瘤篩查未見異常,血清及腦脊液細胞內抗原抗體CV2/CRMP5 PNMA2(Ma2/Ta) Ri Yo Hu Amphiphysin均為陰性(北京協和醫院),患者無神經系統副腫瘤綜合征診斷證據。長期隨訪定期篩查潛在的腫瘤,對疾病的預后十分重要。
患者經過靜點丙種球蛋白、口服潑尼松片等治療后肌肉蠕動癥狀消失,進一步支持病因為自身免疫功能障礙的判斷。但針對LGI1抗體引起周圍神經高興奮性作用機制?自身免疫性甲狀腺抗體升高的病理生理環境下更易誘發抗LGI1抗體相關的腦炎?這些疑問需要我們更多的關注和更長的時間去研究,發現,解答。
猜你喜歡
中老年保健(2021年3期)2021-08-22 06:50:04
天津醫科大學學報(2021年2期)2021-03-29 05:31:08
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
昆明醫科大學學報(2020年12期)2021-01-26 00:44:04
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
豬業科學(2018年8期)2018-09-28 01:27:38
中成藥(2017年8期)2017-11-22 03:18:47
川北醫學院學報(2015年5期)2015-12-05 08:22:29
