替代對(duì)照品法測(cè)定復(fù)方甘草口服溶液中甘草苷和甘草酸含量
2020-12-13 10:04:00魏長(zhǎng)勇余敏靈
中國(guó)藥業(yè) 2020年23期
曾 芳,魏長(zhǎng)勇,余敏靈
(四川省樂(lè)山市食品藥品檢驗(yàn)檢測(cè)中心,四川 樂(lè)山614099)
復(fù)方甘草口服溶液是由甘草流浸膏、愈創(chuàng)甘油醚、復(fù)方樟腦酊等制成的復(fù)方制劑,鎮(zhèn)咳祛痰功效良好。2015年版《中國(guó)藥典(二部)》中未對(duì)甘草苷含量進(jìn)行控制,本研究中以甘草酸銨為對(duì)照品采用高效液相色譜(HPLC)法進(jìn)行甘草酸含量測(cè)定[1]。甘草酸銨易吸潮、價(jià)格貴,參考甘草、甘草流浸膏質(zhì)量標(biāo)準(zhǔn)和相關(guān)文獻(xiàn)[2-9],以橙皮苷為對(duì)照品替代甘草苷、甘草酸銨,采用替代對(duì)照品法,以色譜峰保留時(shí)間為輔助,并采用紫外光譜特征和純度檢查因子作定性鑒別;在定量分析中,通過(guò)測(cè)定橙皮苷和被測(cè)定物質(zhì)的相對(duì)校正因子,并運(yùn)用校正因子計(jì)算甘草苷和甘草酸在制劑中的含量。現(xiàn)報(bào)道如下。
1 儀器與試藥
1.1 儀器
1260型高效液相色譜儀(包括DAD檢測(cè)器,Open-LAB CDS工作站,美國(guó)Agilent公司);PE-A10型高效液相色譜儀(包括DAD檢測(cè)器,珀金埃爾默公司);Ultimate 3000型高效液相色譜儀(包括DAD檢測(cè)器,賽默飛世爾公司);XSE205型電子天平(梅特勒-托利多公司,精度為十萬(wàn)分之一);ULUP-Ⅱ-10T型優(yōu)普系列超純水器(成都超純科技有限公司)。
1.2 試藥
甘草苷對(duì)照品(批號(hào)為111610-201607,含量為93.1%),甘草酸銨對(duì)照品(批號(hào)為110731-201720,含量為97.7%),橙皮苷對(duì)照品(批號(hào)為110721-201818,含量為96.2%),均購(gòu)自中國(guó)食品藥品檢定研究院;甘草酸單銨鹽A原料藥(批號(hào)為1707003,含量為67.2%,北京凱因科技股份有限公司);復(fù)方甘草口服溶液(四川峨嵋山藥業(yè)股份有限公司,批號(hào)分別為20160701,20160702,20160703,20160704,20160705,20160706);乙腈為色譜純,磷酸、甲醇、三乙胺均為分析純;復(fù)方樟腦酊、愈創(chuàng)甘油醚、甘油、濃氨溶液,均購(gòu)自四川峨嵋山藥業(yè)股份有限公司。
2 方法與結(jié)果
2.1 溶液制備
對(duì)照品溶液:稱取甘草苷對(duì)照品17.84 mg,置100 mL容量瓶中,加80%甲醇溶解,并定容至100 mL,作為貯備溶液A;稱取橙皮苷對(duì)照品64.12 mg,置250 mL容量瓶中,加甲醇溶解,并定容至250 mL,作為貯備溶液B;稱取甘草酸銨對(duì)照品11.08 mg,置10 mL容量瓶中,加80%甲醇溶解,并定容至10 mL,作為貯備溶液C。分別量取貯備溶液A和貯備溶液B各1.00 mL、貯備溶液C 2.00 mL,置10 mL容量瓶中,加80%甲醇溶液,并定容至10 mL,作為混合對(duì)照品溶液。
供試品溶液:取樣品(批號(hào)為20160701)4mL,置50mL容量瓶中,加80%甲醇溶液,并定容至50 mL,即得。
樣品-橙皮苷混合溶液:另取樣品(批號(hào)為20160701)4 mL,置50 mL容量瓶中,加入貯備溶液B 5.00 mL,用80%甲醇定容至50 mL,即得。
陰性對(duì)照品溶液:取復(fù)方樟腦酊18 mL、愈創(chuàng)甘油醚0.5 g、甘油12 mL、濃氨溶液適量,加水至100 mL,搖勻,作為陰性樣品溶液;取上述溶液4 mL,置50 mL容量瓶中,加80%甲醇定容至50 mL,作為陰性對(duì)照品溶液;以80%甲醇作為空白溶液。
2.2 測(cè)定波長(zhǎng)選擇
提取混合對(duì)照品溶液和樣品-橙皮苷混合溶液色譜圖中甘草酸銨、甘草苷、橙皮苷色譜峰的紫外光譜圖,各組分在252 nm(甘草酸銨)、276 nm(甘草苷)、284 nm(橙皮苷)波長(zhǎng)處有最大吸收(見(jiàn)圖1),故選擇252,276,284 nm分別作為甘草酸銨、甘草苷、橙皮苷的測(cè)定波長(zhǎng)。
2.3 色譜條件
色譜柱:Welch Ultimate C18柱(250 mm×4.6 mm,5μm);流動(dòng)相:0.1%三乙胺(A,用磷酸調(diào)節(jié)pH至2.5),流動(dòng)相B為乙腈,梯度洗脫(洗脫程序見(jiàn)表1);檢測(cè)波長(zhǎng):252 nm(甘草酸銨)、276 nm(甘草苷)、284 nm(橙皮苷);流速:1.0 mL/min;柱溫:30℃;進(jìn)樣量:10μL。
2.4 方法學(xué)考察
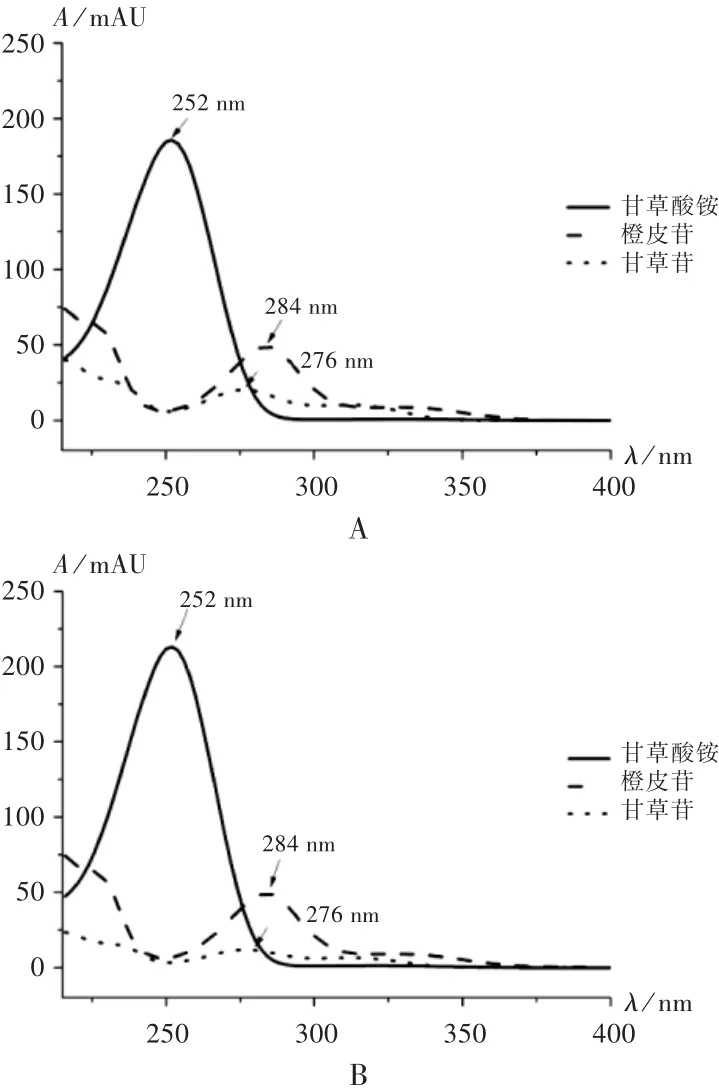
圖1紫外光譜圖

表1流動(dòng)相梯度洗脫程序(%)
專屬性試驗(yàn):取2.1項(xiàng)下空白溶液、陰性對(duì)照品溶液、混合對(duì)照品溶液、供試品溶液、樣品-橙皮苷混合溶液各10μL,按擬訂色譜條件分別注入色譜儀中,測(cè)定,色譜圖見(jiàn)圖2。在284,276,252 nm為檢查波長(zhǎng)的色譜圖中,混合對(duì)照品溶液各組分主峰保留時(shí)間相同的位置上,樣品-橙皮苷混合溶液有相應(yīng)色譜峰,各色譜峰分離度均大于1.5;供試品溶液有與混合對(duì)照品溶液甘草酸銨和甘草苷保留時(shí)間一致的色譜峰,而無(wú)與混合對(duì)照品溶液橙皮苷保留時(shí)間一致的色譜峰;陰性對(duì)照品溶液和空白溶液均無(wú)在與混合溶液保留時(shí)間一致的色譜峰。與混合對(duì)照品溶液保留時(shí)間一致的樣品-橙皮苷混合溶液色譜圖中,甘草酸銨、甘草苷、橙皮苷紫外光譜最大吸收波長(zhǎng)分別為252,276,284 nm。樣品-橙皮苷混合溶液色譜圖中,匹配因子分別為甘草酸銨999.928/1 000,甘草苷999.911/1 000,橙皮苷999.954/1 000。
精密度試驗(yàn):取2.1項(xiàng)下混合對(duì)照品溶液,按擬訂色譜條件重復(fù)進(jìn)樣6次,以每次甘草酸銨、橙皮苷、甘草苷測(cè)定的峰面積計(jì)算。結(jié)果的RSD分別為0.35%,0.24%,0.31%(n=6),表明儀器精密度良好。
重復(fù)性試驗(yàn):精密量取樣品(批號(hào)為20160701)4 mL,依法制備供試品溶液共6份,按擬訂色譜條件進(jìn)樣。結(jié)果甘草酸銨、橙皮苷、甘草苷峰面積的RSD分別為0.68%,0.22%,0.70%(n=6),表明方法重復(fù)性好。
穩(wěn)定性試驗(yàn):取重復(fù)性試驗(yàn)項(xiàng)下制備的供試品溶液10μL,分別于室溫下放置0,2,4,6,8,12,24 h時(shí),按擬訂色譜條件進(jìn)樣測(cè)定。結(jié)果甘草酸銨、橙皮苷、甘草苷峰面積的RSD分別為0.28%,0.33%,0.26%(n=7),表明供試品溶液在室溫放置24 h內(nèi)穩(wěn)定。

圖2高效液相色譜圖
加樣回收試驗(yàn):稱取甘草酸單銨鹽A(批號(hào)為1707003,含量為67.2%)334.82 mg,置100 mL容量瓶中,用80%甲醇溶解并定容至100 mL,作為甘草酸單銨鹽A貯備液;取樣品(批號(hào)為20160701,每1 mL樣品中含甘草苷0.162 8 mg、甘草酸銨2.893 1 mg)2 mL,置50 mL容量瓶中,精密加入貯備溶液A 2 mL、甘草酸單銨鹽A貯備液2 mL、貯備溶液B 5 mL,用80%甲醇定容至50 mL,得供試品溶液,平行制備6份。取10μL,按擬訂色譜條件進(jìn)樣分析,按Cm=(Am×Cs)/(f×As)計(jì)算,Cm(μg)為回收率供試品溶液中甘草酸銨或甘草苷進(jìn)樣量,Am為回收率供試品溶液中甘草酸銨或甘草苷的峰面積,Cs(μg)為回收率供試品溶液中橙皮苷進(jìn)樣量,Cs為回收率供試品溶液中橙皮苷峰面積。結(jié)果見(jiàn)表2。
2.5 相對(duì)校正因子(f)值測(cè)定
取2.1項(xiàng)下混合對(duì)照品溶液2,5,10,15,20,25,30μL按擬訂色譜條件分別注入色譜儀,校正截距為0,以進(jìn)樣量(m,μg)為橫坐標(biāo)、峰面積(A)為縱坐標(biāo)進(jìn)行線性回歸,以甘草酸銨和甘草苷曲線斜率分別除以橙皮苷曲線斜率計(jì)算f值,并以3臺(tái)不同液相色譜儀測(cè)定的平均值作為相對(duì)校正因子。結(jié)果甘草酸銨、橙皮苷、甘草苷線性回歸方程分別為A=803.233 3m,A=1 786.872 0m,A=1 932.570 7m,r分別為0.999 3,0.999 2,0.999 5;線 性 范 圍 分 別 為0.443 0~6.495 1μg,0.049 3~0.740 2μg,0.033 2~0.498 3μg。甘草苷相對(duì)校正因子fA為1.081 4(RSD=0.25%,n=3),甘草酸銨相對(duì)校正因子fB為0.449 2(RSD=0.22%,n=3)。
2.6 相對(duì)校正因子耐受性與相對(duì)保留時(shí)間(tR)考察
取2.1項(xiàng)下混合對(duì)照品溶液,以3臺(tái)不同高效液相色譜儀、不同柱溫(31.0,30.0,29.0℃)、不同測(cè)定波長(zhǎng)(282/274/250 nm,283/275/251 nm,284/276/252 nm,285/277/253nm,286/278/254nm)、不同流動(dòng)相pH(2.9,2.7,2.5)、不同流速(1.005,1.000,0.995 mL/min)的條件進(jìn)行測(cè)定,按2.5項(xiàng)下方法計(jì)算f值,以及橙皮苷色譜峰分別與甘草苷色譜峰和甘草酸銨色譜峰保留時(shí)間的比值。結(jié)果見(jiàn)表3,表明不同液相色譜儀、柱溫流動(dòng)相pH、流速對(duì)tR無(wú)顯著影響。
2.7 紫外光譜穩(wěn)定性考察
取2.1項(xiàng)下混合對(duì)照品溶液,按2.6項(xiàng)下方法(除測(cè)定波長(zhǎng)外)考察,提取橙皮苷、甘草酸銨、甘草苷紫外光譜圖。結(jié)果橙皮苷在(284±2)nm、甘草酸銨在(252±2)nm、甘草苷在(276±2)nm波長(zhǎng)處均有最大吸收;各成分紫外光譜圖與各自對(duì)照品建立的紫外圖庫(kù)相比較,匹配因子均大于999.9/1 000。
2.8 樣品含量測(cè)定
取樣品4 mL,按2.1項(xiàng)下方法制備樣品-橙皮苷混合溶液和供試品溶液。取樣品-橙皮苷混合溶液和供品試溶液10μL,按2.3項(xiàng)下色譜條件,進(jìn)樣測(cè)定,記錄色譜圖,并按回收試驗(yàn)項(xiàng)下公式計(jì)算甘草酸銨和甘草苷進(jìn)樣量,通過(guò)進(jìn)樣量計(jì)算樣品中甘草酸和甘草苷含量,測(cè)定結(jié)果采用配對(duì)t檢驗(yàn),與外標(biāo)法測(cè)定數(shù)據(jù)無(wú)顯著差異(P>0.05)。結(jié)果見(jiàn)表4。

表2加樣回收試驗(yàn)結(jié)果(n=6)

表3甘草苷和甘草酸銨相對(duì)校正因子耐受性試驗(yàn)和相對(duì)保留時(shí)間考察結(jié)果

表4樣品含量測(cè)定結(jié)果(mg/mL)
3 討論
供試品溶液中定量加入替代對(duì)照品作為供試品溶液,替代對(duì)照品測(cè)定波長(zhǎng)按試驗(yàn)設(shè)計(jì)的色譜條件,在未加入替代對(duì)照品的供試品溶液的色譜圖中,不應(yīng)有與替代對(duì)照品溶液保留時(shí)間一致的色譜峰。
由于供試品溶液雜質(zhì)成分較多,對(duì)色譜柱有較高要求,未考察不同色譜柱相對(duì)保留時(shí)間的影響。參考國(guó)家藥品監(jiān)督管理局發(fā)布的國(guó)家藥品標(biāo)準(zhǔn)YBH06492018,建議采用Welch Ultimate C18柱(250 mm×4.6 mm,5μm),本試驗(yàn)中采用以相對(duì)保留時(shí)間為輔助,各組分色譜峰提取紫外光譜圖和對(duì)照品色譜峰所建立的紫外純度檢測(cè)圖庫(kù)比較為主的定性方法。由于工作站所建立的圖庫(kù)只能應(yīng)用于同類色譜儀,故使用局限性較大。
替代對(duì)照品內(nèi)加入法不需要單獨(dú)制備與測(cè)定對(duì)照品溶液,可縮短試驗(yàn)時(shí)間,簡(jiǎn)化檢驗(yàn)程序。可同時(shí)進(jìn)樣測(cè)定待測(cè)組分和對(duì)照品,可降低儀器噪音和操作過(guò)程的影響,結(jié)果準(zhǔn)確,相對(duì)保留時(shí)間穩(wěn)定。

- 中國(guó)藥業(yè)的其它文章
- 有毒中藥鬧羊花的現(xiàn)代研究進(jìn)展*
- 鹽酸舍曲林片聯(lián)合慢性應(yīng)激治療抑郁障礙的療效與安全性評(píng)價(jià)*
- 帕羅西汀治療產(chǎn)后抑郁癥的療效及對(duì)神經(jīng)內(nèi)分泌功能的影響*
- 2010年至2019年國(guó)內(nèi)子宮肌瘤應(yīng)用曼月樂(lè)臨床綜合評(píng)價(jià)*
- 地西他濱聯(lián)合HAG方案治療骨髓增生異常綜合征療效評(píng)價(jià)*
- 基于FAERS的二肽基肽酶-4抑制劑相關(guān)自身免疫性大皰性皮膚病事件數(shù)據(jù)挖掘研究*