羥基自由基對沸石分子篩合成影響的研究
2020-11-08 10:10:04張紅丹齊玉程鵬
應用化工 2020年10期
關鍵詞:沸石
張紅丹,齊玉,程鵬
(沈陽師范大學 化學化工學院 能源與環境催化研究所,遼寧 沈陽 110034)
沸石分子篩材料在吸附、分離、離子交換、非均相催化和精細化工領域發揮了極其重要的作用[1-6]。大多數沸石分子篩材料是在水熱條件下合成的,但這是一個高能耗、污染多、低效率的過程[7]。所以,研究人員一直致力于綠色合成方法的開發,相繼開發出晶種法[8]、微波輔助合成法[9]、無有機模板劑合成法[10]、無溶劑合成法[11]、離子液體合成法等[12]。近期,于吉紅教授使用羥基自由基加速了多種沸石分子篩的晶化過程[13-14]。本課題組也通過預處理,使分子篩的晶種本身產生自由基,加速了沸石分子篩的合成[15]。羥基自由基的發現,為今后沸石分子篩合成體系的發展提供了更廣泛的思路。
1 實驗部分
1.1 試劑與儀器
九水硅酸鈉、偏鋁酸鈉、氫氧化鈉、過硫酸鈉均為分析純;BMPO(自由基捕獲劑),ENZO生命科學;去離子水。
Ultima IV 型X射線衍射儀;JES-FA200 型電子順磁共振譜儀;JSM-6510F 型電子顯微鏡;HC-3018R 型高速冷凍離心機。
1.2 方鈉石的合成
九水硅酸鈉(18.33 g)與去離子水(100.05 g)混合攪拌至澄清,得到A液;偏鋁酸鈉(3.51 g)、氫氧化鈉(42.21 g)與去離子水(100.05 g)混合攪拌至澄清并降至室溫,得到B液;將B液滴加到A液中,繼續攪拌15 min。而后,分別將10 mL反應液轉移到18個反應釜中。將這些反應釜分成2份,其中一份不做處理,另一份每個釜中加入0.5 mL 0.42 mol/L的過硫酸鈉溶液。將其放入80 ℃烘箱中晶化,分不同的晶化時間(1,1.5,2,2.5,3,3.5,4,5,6 h)取出,快速冷卻。將晶化后的溶液進行固液離心分離,留取底部固體,并使用去離子水再次分散,如此3次,最后得到固體產物,在70 ℃烘箱中干燥整晚。
2 結果與討論
2.1 XRD分析
將得到的固體產物進行X-射線衍射表征,結果見圖1。
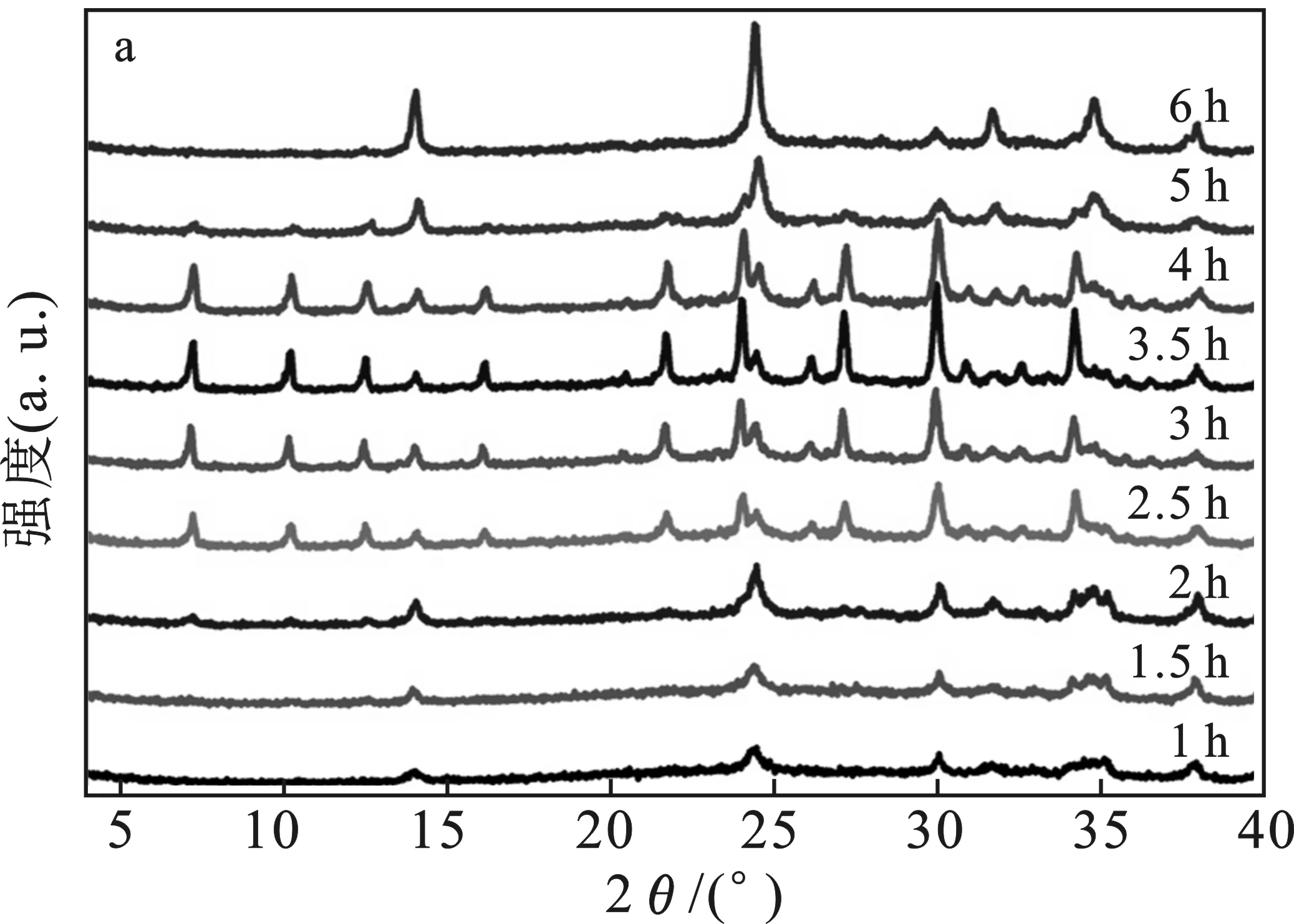
由圖1a可知,不加入過硫酸鈉溶液時,前期的衍射峰中主要是前驅體的特征峰,伴有極少量的NaA沸石的特征峰。隨著晶化時間的延長,NaA沸石的特征峰強度逐漸增強,同時出現了少量方鈉石的特征峰,此時產物是兩相共存的狀態,NaA沸石為主相。5 h后,方鈉石變為主相,固體產物中NaA沸石含量極少,直至6 h,固體產物完全變成了方鈉石。
由圖1b可知,加入過硫酸鈉溶液后,前驅體的峰僅存在晶化1.5 h之前,2~3 h就已經產生了晶化度很好的方鈉石,但是在其中伴隨產生了極少量的NaA沸石。晶化3.5 h后,固體產物就已經變成晶化度良好的方鈉石材料。
2.2 EPR分析
為了探究過硫酸鈉溶液是否確實產生了羥基自由基,以BMPO作為自由基捕獲劑,對過硫酸鈉溶液和空白溶液進行了電子順磁共振表征,結果見圖2。
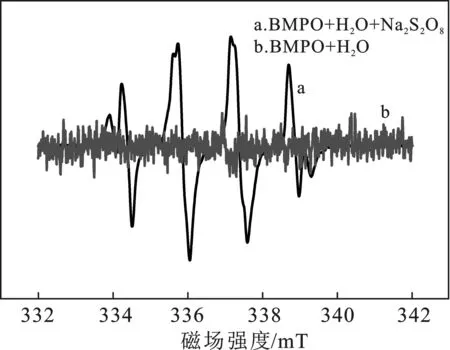
圖2 以BMPO為捕獲劑,過硫酸鈉溶液與純水的電子順磁共振譜圖Fig.2 EPR spectra of sodium persulfate solution and H2O containing the spin-trapping agent of BMPO
由圖2可知,純水溶液產生了極弱強度的羥基自由基的特征峰,而過硫酸鈉溶液則產生了更強的羥基自由基的信號強度,說明溶液中加入了過硫酸鈉后,額外產生了大量的羥基自由基。
2.3 SEM分析
圖3展示了不加入過硫酸鈉的固體產物的掃描電子顯微鏡照片。

圖3 不加入過硫酸鈉不同晶化時間的固體產物掃描電子顯微鏡照片Fig.3 SEM images of the solid products of different crystallized time in the absence of sodium persulfatea.2.5 h;b.3 h;c.3.5 h;d.4 h;e.5 h;f.6 h
由圖3可知,晶化3 h前,產物中仍然存在無定形的大塊固體。隨著晶化時間的延長,產物逐漸變成了正方體晶體(NaA沸石)。5 h后,正方體逐漸溶解,說明NaA沸石逐漸轉化為另一種沸石。從X-射線衍射的結果可知,其轉化為方鈉石。
圖4展示了加入過硫酸鈉的固體產物的掃描電子顯微鏡照片。

圖4 過硫酸鈉存在下不同晶化時間的固體產物掃描電子顯微鏡照片Fig.4 SEM images of the solid products of differentcrystallized time in the presence of sodium persulfate a.1.5 h;b.2 h;c.2.5 h;d.3 h;e.3.5 h;f.4 h;g.5 h;h.6 h
由圖4可知,2 h前,除了小球狀晶體外,還存在一定的大塊無定形材料。2.5 h后,即出現了沒有其他雜質的小球狀晶體,大小均一,分散性好。
3 結論
使用羥基自由基輔助合成沸石分子篩的過程中發現,羥基自由基可以提高沸石分子篩的相選擇性,得到更純、晶化度更好的沸石分子篩,同時可以提高合成沸石分子篩的效率。
猜你喜歡
山西化工(2024年2期)2024-03-20 07:33:10
云南化工(2021年10期)2021-12-21 07:33:24
煤氣與熱力(2021年9期)2021-11-06 05:22:56
化學工業與工程(2021年5期)2021-11-03 03:43:56
湖南飼料(2021年3期)2021-07-28 07:06:06
天然氣化工—C1化學與化工(2019年6期)2019-02-18 07:06:02
電鍍與環保(2017年6期)2018-01-30 08:33:35
中國非金屬礦工業導刊(2015年5期)2015-12-22 06:26:12
石油化工(2015年9期)2015-08-15 00:43:05
應用化工(2014年1期)2014-08-16 13:34:08
