介孔TiO2晶須-γ-Al2O3復合載體催化劑的制備及對二苯并噻吩的加氫脫硫性能
2020-09-27 09:11:32楊祝紅花澤林李力成
石油學報(石油加工) 2020年4期
岳 凡,李 蒙,楊祝紅,花澤林,李 龍,李力成
(1.南京工業(yè)大學 化工學院,江蘇 南京 210009;2.南京林業(yè)大學 化學工程學院,江蘇 南京 210037)
近年來,環(huán)境問題逐漸受到大眾的廣泛關注,中國出臺的相關法規(guī)對污染排放控制也隨之愈發(fā)嚴格,其中GB 19147—2016《車用柴油》標準要求燃料油中的硫質(zhì)量分數(shù)低于10 μg/g,這對目前工業(yè)上廣泛使用的油品加氫脫硫技術(shù)(HDS)提出了更高的要求,而催化劑性能的優(yōu)劣是決定該技術(shù)的核心。
在眾多催化劑調(diào)控技術(shù)中,開發(fā)新型載體材料是提升加氫脫硫催化劑性能較為簡略的有效途徑之一。目前最為廣泛應用的加氫脫硫催化劑載體是γ-Al2O3,其具有高比表面積、高熱穩(wěn)定性和良好的孔結(jié)構(gòu)等特點[1-2]。但是γ-Al2O3與活性組分間有強相互作用,容易形成尖晶石結(jié)構(gòu)的穩(wěn)定物質(zhì),這類物質(zhì)不利于硫化形成催化活性中心[3],導致所制得的催化劑脫硫性能下降,因此,僅以γ-Al2O3為載體的催化劑將難以應對更加嚴峻的挑戰(zhàn)。為了進一步提高催化劑的加氫脫硫性能,大量技術(shù)人員研究了其他類型的催化劑載體材料,如Zhang等[4]通過水熱法制備出鈦硅沸石納米棒(TS-1),負載鈷、鉬制備了CoMo/TS-1催化劑,經(jīng)研究發(fā)現(xiàn),TS-1骨架中的Ti物種可以增強Mo與TS-1間的相互作用,有助于暴露更多有利于加氫的活性位點,進而提高催化劑的加氫脫硫性能。Cortes-Jacome等[5]采用水熱法制備出高比表面積的氧化鈦納米管,并以此為載體制備出高分散的CoMoS催化劑,可表現(xiàn)出良好的脫硫性能。另外,Huang等[6]以四正丁基鈦酸為鈦源,采用溶膠-凝膠法制備TiO2-Al2O3復合載體,負載鎳、鉬制備出具有高活性的加氫脫硫催化劑。
由上可見,Ti的引入能夠有效避免純γ-Al2O3載體催化劑上存在的問題,通過將TiO2與γ-Al2O3載體進行復合,可以有效地發(fā)揮TiO2與γ-Al2O3各自的優(yōu)勢,進而制備出優(yōu)異的加氫脫硫催化劑。然而,常規(guī)TiO2作為載體存在比表面積小、機械強度差等缺點,雖然用溶膠-凝膠等[7-8]方法能夠制備出高比表面積的TiO2,但是制備成本高、工業(yè)化困難等缺點限制了其在工業(yè)方面的應用,將其進一步與γ-Al2O3進行復合,會對所制得的復合載體的孔結(jié)構(gòu)、兩者間相互復合強度以及機械性能等因素產(chǎn)生負面影響。筆者所在課題組前期[9]從二鈦酸鉀(K2Ti2O5)晶須出發(fā),成功制得比表面積大于 139 m2/g 的介孔TiO2晶須材料,所制得的TiO2成型載體抗破碎強度大于12 N/mm。劉金龍等[10]、朱銀華等[11]、Li等[12]以TiO2為載體制備HDS催化劑,均顯示出優(yōu)異的催化活性。在此基礎上,溫廣明等[13]開展介孔TiO2晶須與γ-Al2O3的復合制備研究,可以有效克服常規(guī)TiO2與γ-Al2O3復合所存在的不足,并發(fā)現(xiàn)介孔TiO2晶須在復合載體中的最佳質(zhì)量分數(shù)為5%,初步負載活性組分后,所得催化劑可表現(xiàn)出優(yōu)異的加氫脫硫性能。
然而,已有文獻[14-15]對不同載體進行活性組分負載過程對比考察的結(jié)果表明,催化劑性能受到載體的表面性質(zhì)、孔結(jié)構(gòu)、晶相等多方面因素影響,所得到的實驗結(jié)果難以直接借鑒到其他類型的催化劑載體,故針對新型載體材料需要重新開展活性組分負載研究。因此,筆者對新型的介孔TiO2晶須-γ-Al2O3復合載體開展活性組分負載量的系統(tǒng)性研究,通過一系列技術(shù)手段對所制備的催化劑進行結(jié)構(gòu)表征,并以DBT作為模型反應物對催化劑的性能進行評價。
1 實驗部分
1.1 試劑和原料
鉬酸銨((NH4)6Mo7O24·4H2O)、二硫化碳(CS2)、十氫萘(C10H18),均為分析純,國藥集團化學試劑有限公司產(chǎn)品;二苯并噻吩(C12H8S,質(zhì)量分數(shù)99%),麥克林試劑有限公司產(chǎn)品;硝酸(HNO3,質(zhì)量分數(shù)69%),國藥集團化學試劑有限公司產(chǎn)品。介孔TiO2晶須,自制,制備方法參考文獻[7];擬薄水鋁石,工業(yè)級,淄博萬霖化工科技有限公司產(chǎn)品;TiO2(比表面積11.9 m2/g),分析純,國藥集團化學試劑有限公司產(chǎn)品。
1.2 載體和催化劑的制備
采用常規(guī)的溶膠-凝膠法制備介孔TiO2晶須-γ-Al2O3復合載體,具體步驟如下:稱取10 g擬薄水鋁石放入裝有200 mL去離子水的三角燒瓶中,攪拌混勻;將三口燒瓶置于油浴鍋中加熱至70 ℃并保持恒溫,緩慢滴加稀硝酸,將混合物體系pH值調(diào)至1左右,加入0.5 g介孔TiO2晶須;繼續(xù)攪拌6 h,期間每隔0.5 h補充適量的稀硝酸維持體系pH=1;結(jié)束后,緩慢滴加適量氨水,將體系pH值調(diào)至9左右,形成復合膠狀物,陳化3 h后取出,用去離子水將其洗至pH=7,經(jīng)抽濾、烘干、550 ℃ 焙燒2 h后得到介孔TiO2晶須-γ-Al2O3復合載體,記為TiO2-Al2O3。以常規(guī)TiO2為鈦源,采用同樣的方法制備TiO2質(zhì)量分數(shù)為5%的復合載體,記為c-TiO2-Al2O3。
以(NH4)6Mo7O24·4H2O作為鉬源配制前驅(qū)體溶液,通過等體積浸漬法負載活性組分。將一定量前驅(qū)體溶液加入到載體中,經(jīng)攪拌、陳化、干燥和500 ℃焙燒2 h后得到不同MoO3含量的介孔TiO2晶須-γ-Al2O3復合載體催化劑,記為x-MoO3/TiO2-Al2O3,其中x為MoO3的質(zhì)量分數(shù)。另外,采用同樣的方法制備純γ-Al2O3為載體及c-TiO2-Al2O3為載體MoO3質(zhì)量分數(shù)為20%的催化劑作為參比樣,分別記為20%MoO3/γ-Al2O3和20%MoO3/c-TiO2-Al2O3。
1.3 催化劑表征
采用美國Micromeritics公司TristarII 3020M型比表面孔隙吸附儀分析各個催化劑的孔結(jié)構(gòu),測試前,樣品在150 ℃下脫氣處理6 h,以N2作為吸附質(zhì)在液氮(-196 ℃)下進行測定。樣品的微觀形貌結(jié)構(gòu)分析以及EDX測試采用日立公司生產(chǎn)的Hitachi S-4800場發(fā)射掃描電鏡(FESEM)進行,操作電壓為5 kV。硫化后催化劑的形貌分析在JEOL JEM-2100透射電子顯微鏡上進行,通過TEM顯微照片確定的顆粒尺寸來統(tǒng)計評估MoS2顆粒的尺寸信息。樣品的晶體結(jié)構(gòu)分析采用德國Bruke公司的D8 Adavance型X射線衍射儀,CuKα輻射源,管電流為100 mA,管電壓為40 kV,掃描范圍2θ=5°~80°,掃描步長為0.02°,掃描速率為20 s/step。樣品的拉曼光譜分析在配備CCD相機探測器的Horiba HR 800光譜儀上進行,采用波長為 514 nm Ar/Kr離子激光器作為激發(fā)光源,物鏡放大倍數(shù)為50倍,樣品激光功率不超過5 mV。氫氣程序升溫還原(H2-TPR)實驗在天津先權(quán)工貿(mào)發(fā)展有限公司的TP-5000型多用吸附儀上進行。硫化狀態(tài)催化劑的X-射線光電子能譜分析在ESCALAB 250光譜儀(美國Thermo Fisher Scientific公司產(chǎn)品)上進行,AlKαX射線激發(fā)源,功率300 W。
1.4 催化劑活性評價
催化劑的加氫脫硫性能在固定床微反應裝置上進行評價。將0.33 g催化劑放置在反應器的恒溫段,反應前將催化劑置于3%(質(zhì)量分數(shù))CS2/十氫萘溶液中,在溫度300 ℃、H2壓力2 MPa、H2流量20 mL/min、硫化液流量0.033 mL/min條件下預硫化處理6 h。預硫化結(jié)束后,將硫化液切換成1%(質(zhì)量分數(shù))DBT/十氫萘反應液,在溫度300 ℃、H2壓力2 MPa、氫/油體積比600、體積空速6 h-1的條件下進行反應。預反應5 h穩(wěn)定后開始采集第一個產(chǎn)物,之后每隔1 h采集一次反應產(chǎn)物。反應產(chǎn)物采用山東魯南瑞虹化工儀器有限公司SP-6890 型氣相色譜儀進行檢測。DBT轉(zhuǎn)化率通過公式(1)計算。
(1)
DBT的加氫脫硫途徑主要有兩種[12]:直接脫硫(DDS)產(chǎn)物為聯(lián)苯(BP),氫化脫硫(HYD)產(chǎn)物為環(huán)己基苯(CHB)。DDS選擇性根據(jù)公式(2)計算。
(2)
式(1)和(2)中,win和wout分別為原料和產(chǎn)物中DBT的質(zhì)量分數(shù),%;w為產(chǎn)物中聯(lián)苯的質(zhì)量分數(shù),%。
2 結(jié)果與討論
2.1 催化劑的物性表征結(jié)果
2.1.1 XRD分析
圖1是TiO2-Al2O3復合載體和不同MoO3含量MoO3/TiO2-Al2O3催化劑以及20%MoO3/c-TiO2-Al2O3催化劑的XRD譜圖。由圖1可見,TiO2-Al2O3復合載體樣品在2θ為46.0°和67.0°處的衍射峰對應于γ-Al2O3相[16]的特征峰,而在2θ為25.3°和48.0°兩處微弱的衍射峰對應于銳鈦礦相TiO2[17]的特征峰。此外,當MoO3負載質(zhì)量分數(shù)為25%和30%時,在2θ為23.5°處出現(xiàn)明顯的MoO3衍射峰[11],這主要是由于MoO3負載量過高,在載體表面發(fā)生了團聚所引起。當MoO3負載質(zhì)量分數(shù)低于20%時,在MoO3/TiO2-Al2O3催化劑的XRD曲線上沒有出現(xiàn)MoO3衍射峰,表明在該范圍內(nèi)活性金屬在載體表面具有良好的分散性,這有利于催化劑還原和提高反應活性。而20%MoO3/c-TiO2-Al2O3催化劑在2θ為25.3°、37.7°、48.0°、53.8°和55.1°處均出現(xiàn)明顯的銳鈦礦型TiO2的衍射峰[17],并且在2θ為23.5°處出現(xiàn)明顯的MoO3衍射峰。此結(jié)果表明20%MoO3/c-TiO2-Al2O3催化劑中的活性金屬發(fā)生團聚,生成了晶態(tài)MoO3,而MoO3負載質(zhì)量分數(shù)為20%時,20%MoO3/TiO2-Al2O3催化劑中并沒有出現(xiàn)晶態(tài)MoO3的衍射峰。

圖1 TiO2-Al2O3復合載體以及負載不同含量MoO3的催化劑XRD圖譜
2.1.2 比表面積和孔結(jié)構(gòu)分析
圖2為TiO2-Al2O3復合載體以及負載不同含量MoO3的催化劑的N2吸附-脫附等溫線。由圖2(a)可見,所有樣品的N2吸附-脫附等溫線均呈IV型,這表明所有材料均具有介孔結(jié)構(gòu)[18]。圖2(b)的孔徑分布圖顯示,各催化劑以及復合載體的最可幾孔徑主要集中在5 nm左右,進一步證明復合載體催化劑具有介孔結(jié)構(gòu)。
表1為復合載體及其不同MoO3含量催化劑的比表面積、平均孔徑及孔體積數(shù)據(jù)。由表1可知,TiO2-Al2O3復合載體的比表面積為308.7 m2/g,孔體積為0.56 cm3/g,平均孔徑為6.1 nm,具有豐富的孔結(jié)構(gòu)。而常規(guī)TiO2為Ti源的c-TiO2-Al2O3復合載體的比表面積僅為203.4 m2/g,孔體積為0.32 cm3/g,平均孔徑為4.5 nm。負載活性組分MoO3后,所制得的催化劑比表面積和孔體積均有所降低,當MoO3負載質(zhì)量分數(shù)為30%時,MoO3/TiO2-Al2O3催化劑的比表面積和孔體積最低;而20%MoO3/c-TiO2-Al2O3催化劑的比表面積遠小于20%MoO3/TiO2-Al2O3的。負載MoO3后催化劑的比表面積和孔結(jié)構(gòu)數(shù)據(jù)減小是由于金屬負載于載體表面和孔道之中,導致載體部分表面以及小孔孔道被覆蓋或堵塞[19]。

圖2 TiO2-Al2O3復合載體與不同MoO3含量催化劑的N2吸附-脫附曲線和孔徑分布圖
2.1.3 Raman分析
圖3給出了不同MoO3負載量的MoO3/TiO2-Al2O3復合載體催化劑的Raman光譜圖。由圖3可知:所有催化劑在640 cm-1處出現(xiàn)的Raman信號峰對應于銳鈦礦型TiO2的特征峰[20];在865 cm-1處出現(xiàn)的Raman信號峰與低聚體MoOx中橋接Mo—O—Mo的對稱和反對稱伸縮振動有關[21],并且隨著催化劑中Mo含量增加,拉曼振動峰強度先增加后減少;在930~970 cm-1范圍內(nèi)的寬信號峰可能與氧化鉬中的Mo=O的伸縮振動有關[22-23],當MoO3負載量較低時,930~970 cm-1幾乎沒有拉曼峰出現(xiàn),隨著MoO3負載量增加,930~970 cm-1的拉曼信號峰強度明顯增加,表明在催化劑表面形成了更多 Mo=O 的Mo物種,有報道指出這種Mo物種有利于提高催化劑活性[24]。
2.1.4 SEM和TEM分析
圖4是5%MoO3/TiO2-Al2O3、20%MoO3/TiO2-Al2O3和30%MoO3/TiO2-Al2O3催化劑的SEM照片。由圖4可以看出,金屬負載量的增加并沒有對載體的形貌產(chǎn)生大的影響。為了證明氧化狀態(tài)的Mo在載體表面的分散度差異,采用SEM-EDX對 20%MoO3/TiO2-Al2O3和30%MoO3/TiO2-Al2O32個催化劑進行EDX測試,在每個催化劑中隨機取2點進行測試,結(jié)果如圖5所示。由圖5可知,MoO3

圖3 不同MoO3負載量的MoO3/TiO2-Al2O3復合載體催化劑Raman光譜圖
負載質(zhì)量分數(shù)為20%時,催化劑表面所測得的Mo金屬元素的質(zhì)量分數(shù)分別為13.17%和13.04%,Mo含量基本一致,表明MoO3質(zhì)量分數(shù)為20%時在載體表面分散均勻;MoO3負載質(zhì)量分數(shù)為30%時,催化劑表面所測得的Mo元素的質(zhì)量分數(shù)分別為18.12%和19.51%,差別較大,此結(jié)果表明,MoO3質(zhì)量分數(shù)為30%時,活性金屬在載體表面的團簇使局部Mo金屬含量偏高。圖6是新鮮硫化的20%MoO3/TiO2-Al2O3和30%MoO3/TiO2-Al2O3催化劑的TEM照片。TEM結(jié)果表明各催化劑表面的MoS2均呈現(xiàn)層狀結(jié)構(gòu)。通過統(tǒng)計(對150個MoS2進行尺寸統(tǒng)計)和計算[25]得到20%MoO3/TiO2-Al2O3和30%MoO3/TiO2-Al2O3的MoS2片晶的平均長度分別為2.5 nm和2.7 nm,堆疊層數(shù)分別為1.9和2.0。
2.1.5 H2-TPR分析
圖7是不同MoO3含量MoO3/TiO2-Al2O3催化劑的H2-TPR圖譜。由圖7看到,所有催化劑的TPR曲線均在350~500 ℃范圍內(nèi)出現(xiàn)1個明顯的還原峰,這對應于八面體配位的聚合Mo物種從Mo6+到Mo4+的還原過程。Zhou等認為,該類型的Mo物種與載體之間的相互作用弱,易于還原成具有優(yōu)異加氫脫硫性能的活性物種[26]。MoO3負載質(zhì)量分數(shù)低于20%時,催化劑還原峰峰值為475 ℃,當MoO3負載質(zhì)量分數(shù)高于20%時,催化劑的還原溫度略有升高,還原峰的峰值由475 ℃升高至494 ℃,并且在500~600 ℃范圍內(nèi)出現(xiàn)1個小的還

圖4 不同MoO3含量MoO3/TiO2-Al2O3催化劑的SEM照片

圖5 不同MoO3含量MoO3/TiO2-Al2O3催化劑的SEM-EDX譜

圖6 不同MoO3含量MoO3/TiO2-Al2O3催化劑的TEM圖
原峰。據(jù)報道,晶態(tài)MoO3在480、575和665 ℃處出現(xiàn)還原峰[27],而圖1的XRD結(jié)果顯示,MoO3負載質(zhì)量分數(shù)超過20%時催化劑上會出現(xiàn)晶態(tài)MoO3,由此判斷該還原峰為晶態(tài)MoO3的還原峰。圖7中還看到,20%MoO3/γ-Al2O3催化劑的還原峰峰值在506 ℃,而MoO3/TiO2-Al2O3催化劑的還原峰峰值明顯低于506 ℃,表明TiO2的引入能夠有效減弱Mo與Al2O3之間的相互作用,從而促進形成更多的活性相。
2.1.6 XPS分析
圖8是硫化態(tài)的20%MoO3/TiO2-Al2O3催化劑的Mo 3d的XPS圖譜。由圖8可知,硫化后的催化劑主要存在2種價態(tài)的Mo物種,特征峰的結(jié)合能位置分別為232.5和238.6 eV,可歸屬于Mo6+物種[28](Mo VI: (232.5±0.1) eV 3d5/2, (238.6±0.1) eV 3d3/2),特征峰的結(jié)合能位置分別為229 eV和232.1 eV的可歸因于Mo4+物種(Mo IV:(229.0±0.1) eV 3d5/2, (232.2±0.1) eV 3d3/2)[28]。van Haandel等[29]對比研究了硫化前后的Mo物種結(jié)合能變化情況,發(fā)現(xiàn)硫化前催化劑中的Mo物種只在高結(jié)合能位置(232.5 eV和238.6 eV)出現(xiàn)特征峰,經(jīng)硫化后,Mo物種在高結(jié)合能位置和低結(jié)合能(229.0 eV和232.3 eV)位置均出現(xiàn)特征峰,認為這是催化劑中高價態(tài)的Mo物種被還原成低價態(tài)Mo物種所致。同理,圖8中在低結(jié)合能處出現(xiàn)的Mo4+物種信號表明催化劑中Mo物種在硫化過程中發(fā)生還原。通過分峰計算可知20%MoO3/TiO2-Al2O3催化劑的硫化度為32.2%(硫化度計算公式:F=AMo4+/(AMo4++AMo6+)×100%,其中F為硫化度,AMo4+和AMo6+分別代表XPS中Mo4+和Mo6+的特征峰面積),Zhang等[30]在相同硫化條件下發(fā)現(xiàn)MoO3/Al2O3系列催化劑中Mo的硫化度為22%~31%,顯然,本實驗中MoO3/TiO2-Al2O3催化劑的硫化度更高,這可歸因于MoO3/TiO2-Al2O3催化劑中的TiO2促進了MoO3發(fā)生硫化。

圖7 不同MoO3負載量的MoO3/TiO2-Al2O3復合載體催化劑和20%MoO3/γ-Al2O3的H2-TPR譜圖
2.2 不同催化劑樣品的DBT加氫脫硫反應性能評價
不同催化劑的DBT加氫脫硫反應結(jié)果如表2所示。表2表明:MoO3/TiO2-Al2O3催化劑的DBT轉(zhuǎn)化率隨MoO3負載量增加呈先增加后降低的趨勢,當MoO3負載質(zhì)量分數(shù)為20%時,MoO3/TiO2-Al2O3催化劑在當前的反應條件下表現(xiàn)出最佳的加氫脫硫性能,其DBT轉(zhuǎn)化率達到56%;20%MoO3/γ-Al2O3和20%MoO3/c-TiO2-Al2O3的DBT轉(zhuǎn)化率分別為49%和41%,由此可見介孔TiO2晶須-Al2O3復合載體作為脫硫催化劑載體可以表現(xiàn)出比純γ-Al2O3和常規(guī)TiO2為Ti源的復合載體更為優(yōu)異的深度加氫脫硫性能。Ferdous等[31]報道,Ti的引入可以有效促進Mo在Al2O3催化劑表面形成更多活性多鉬氧化物。因此可以解釋介孔TiO2晶須-Al2O3復合載體催化劑的脫硫性能優(yōu)于γ-Al2O3催化劑。常規(guī)TiO2為Ti源的c-TiO2-Al2O3載體催化劑的加氫脫硫活性低于介孔TiO2晶須-Al2O3復合載體催化劑和γ-Al2O3催化劑,可能是由于載體比表面積低,使得Mo金屬在載體表面團聚形成不利于催化活性的晶態(tài)MoO3。而另一方面,由2.1節(jié)表征結(jié)果可知,當MoO3負載量較低時,MoO3能夠在載體表面良好地分散,在該條件下,催化劑的活性位點數(shù)量隨負載量增加也得到了進一步的提升;然而,當負載質(zhì)量分數(shù)高于分散閾值(20%)時,MoO3在載體的分散度有所下降,活性金屬團聚形成了難以還原的晶態(tài)MoO3,進而使得催化劑的DBT轉(zhuǎn)化率下降。
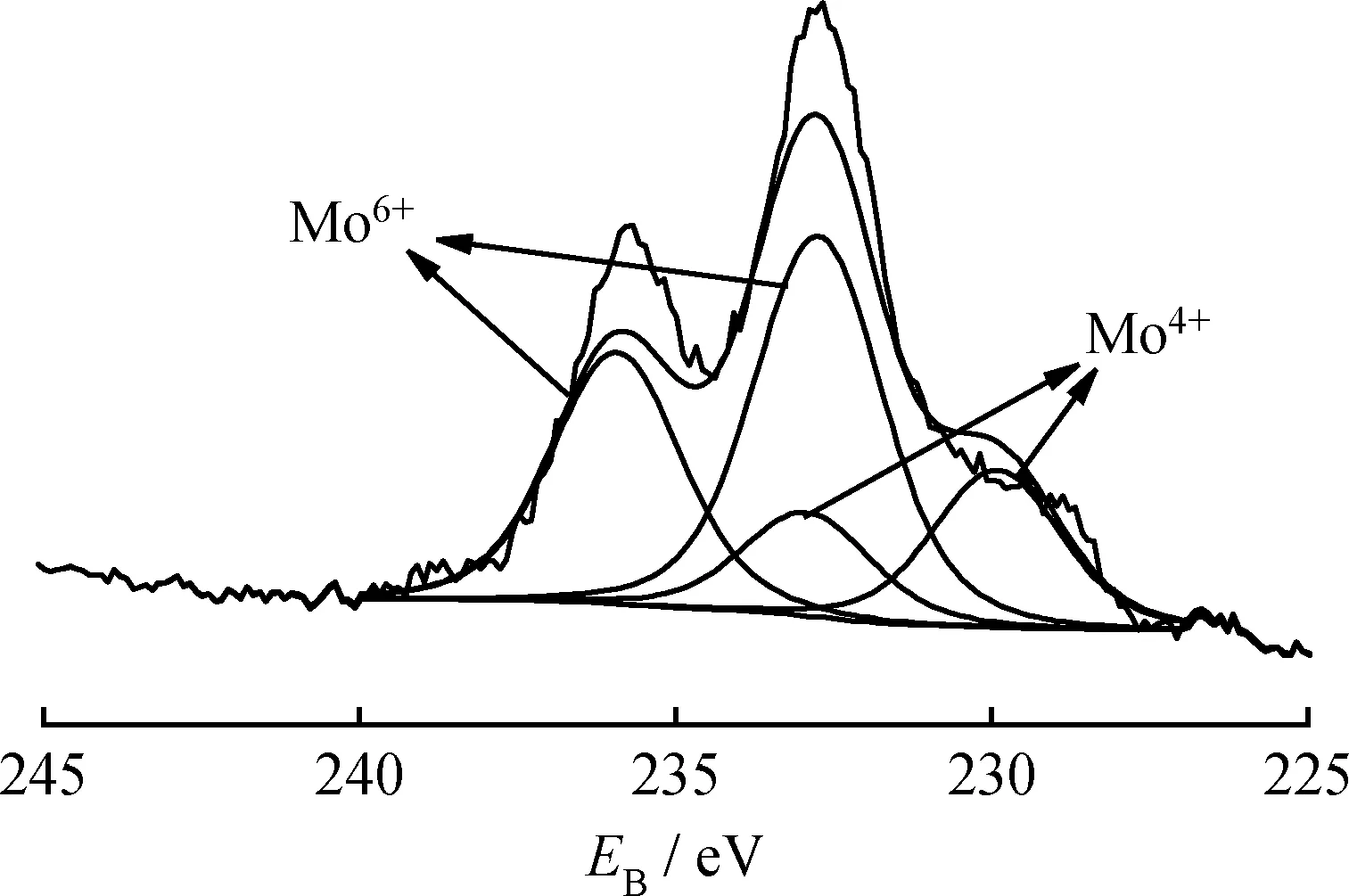
圖8 硫化后20%MoO3/TiO2-Al2O3催化劑的XPS譜圖
由表2可知,不同MoO3負載量的 MoO3/TiO2-Al2O3催化劑的DDS選擇性均在70%左右,表明MoO3負載量的變化沒有對催化劑的DDS選擇性產(chǎn)生明顯的影響。20%MoO3/c-TiO2-Al2O3催化劑的DDS選擇性在57%左右,而20%MoO3/γ-Al2O3催化劑的DDS選擇性僅為46%,明顯低于復合載體催化劑,表明Ti引入使得催化劑更加傾向于DDS路徑。Vazquez-Garrido等[32]和Guevara等[33]的研究結(jié)果同樣表明,TiO2能夠有效減弱活性金屬與Al2O3間相互作用,從而增加催化劑的DDS選擇性。

表2 不同催化劑加氫脫硫反應的DBT轉(zhuǎn)化率和DDS選擇性
3 結(jié) 論
制備了介孔TiO2晶須與γ-Al2O3復合載體(TiO2-Al2O3)及不同MoO3負載量的MoO3/TiO2-Al2O3催化劑,并進行了物性表征和對二苯并噻吩的加氫脫硫反應性能評價,發(fā)現(xiàn)當MoO3負載質(zhì)量分數(shù)為20%時,MoO3在復合載體上分散良好,所制得的20%MoO3/TiO2-Al2O催化劑具有高的比表面積和豐富的介孔結(jié)構(gòu),活性組分易于還原,能夠展示出最佳的加氫脫硫性能,DBT轉(zhuǎn)化率達到56%,優(yōu)于相同條件下純γ-Al2O3為載體的催化劑(49%)。
猜你喜歡
中學生數(shù)理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數(shù)理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
產(chǎn)品可靠性報告(2017年7期)2017-09-05 09:49:12
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
汽車觀察(2016年3期)2016-02-28 13:16:26
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
