2010-2019年某CRO公司藥物臨床試驗項目盲態審核中方案偏離的相關數據分析
2020-09-21 08:46:35董玉君王蓉余秋鈿程國華
中國藥房 2020年17期
關鍵詞:質量管理
董玉君 王蓉 余秋鈿 程國華

摘 要 目的:研究方案偏離對藥物臨床試驗結果的影響,為提高藥物臨床試驗的質量管理水平提供參考。方法:收集廣州某合同研究組織(CRO)公司2010-2019年藥物臨床試驗的盲態數據審核表,分析方案偏離總體特征、“722公告”(指國家食品藥品監督管理總局2015年7月22日發布《關于開展藥物臨床試驗數據自查核查工作的公告》)前后的方案偏離情況以及方案偏離對全分析集(FAS)人群劃分的影響,并提出相應建議。結果:最終納入45個試驗項目,涉及454個中心、14 304例病例和5 562例次方案偏離。最常見的方案偏離類型依次是超窗、違背納入與排除標準、脫落,分別占方案偏離的36.88%、20.71%、18.43%。不同試驗分期和不同藥物類型的項目發生方案偏離的程度無統計學意義(P>0.05),而“722公告”前后不同階段項目發生方案偏離的程度有顯著性差異(P<0.05);“722公告”后的超窗、違背納入與排除標準和服藥依從性的偏離發生率有所升高。發生方案偏離的病例有82.07%可以進入FAS,納入FAS且未進入符合方案集(PPS)的人群占總體偏離人群的53.99%,其中脫落、合并用藥偏離分別占19.51%、4.29%,服藥依從性偏離的人群均未進入PPS。結論:脫落、違背納入與排除標準、超窗是造成臨床試驗方案偏離的主要因素。“722公告”對藥物臨床試驗人員的質量管理意識的提高起到了一定的促進作用。建議應選擇恰當的統計方法控制偏倚,從試驗設計、人員培訓及機構管理建設各方面加強藥物臨床試驗質量管理,減少方案偏離的發生。
關鍵詞 方案偏離;藥物臨床試驗;盲態審核;質量管理
ABSTRACT? ?OBJECTIVE: To study the effects of protocol deviation on the results of clinical trials, and to provide reference for rising the quality management of drug clinical trials. METHODS: Blind data review forms for clinical trials of a contract research organzation (CRO) company in Guangzhou from 2010 to 2019 were collected to analyze general characteristics of protocol deviation, the situation of protocol deviation before and after the “722 announcement” (Announcement on Carrying Out Self-inspection and Verification of Drug Clinical Trial Data issued by CFDA on July 22, 2015) as well as the effects of protocol deviation on full analysis set (FAS) population division. The suggestions were put forward. RESULTS: A total of 45 trials were included, involving 454 centers, 14 304 disease cases and 5 562 cases of protocol deviation. The most common types of protocol deviations were over-window, violation of criteria of the inclusion and exclusion, and drop-out, which accounted for 36.88%, 20.71% and 18.43% respectively. There was no statistical significance in protocol deviation degree of clinical trials with different stages or drug types (P>0.05); there was significant difference in the degree of protocol deviations in clinical trials with different stages before and after the “722 announcement” (P<0.05); the incidence of deviations from over-window, violation of cirteria of the inclusion and exclusion, and medication compliance had increased after the “722 announcement”; 82.07% of cases with protocol deviations could enter FAS, and the population who included in FAS but did not enter per protocol set (PPS) accounted for 53.99% of the total deviation, of which deviations from drop-out and combined medication accounted for 19.51% and 4.29% respectively. All cases with deviation from medication compliance did not enter PPS. CONCLUSIONS: Drop-out, violation of criteria of the inclusion and exclusion, and over-window are the main factors that cause clinical trial protocol deviations. The “722 announcement” played a certain role on improving the quality management awareness of the personnel in drug clinical trial. Appropriate statistical methods should be selected to control bias, and to strengthen the quality management of drug clinical trials and reduce protocol deviations, by paying attention to trial design, staff training, institutional management and construction.
KEYWORDS? ?Protocol deviation; Drug clinical trial; Blind review; Quality management
任何有意或無意偏離或違反《藥物臨床試驗質量管理規范》(GCP)或試驗方案的行為被稱作方案偏離(Protocol deviation)或方案違背(Protocol violation)[1]。當方案偏離積累到某種程度時將會影響試驗的有效性和安全性評價結果。盲態審核是臨床試驗數據管理的關鍵步驟,是數據管理與統計分析銜接的橋梁[2]。試驗結束到揭盲之前,主要研究者、醫學總監、生物統計學家、數據管理員和申辦者會在盲態的情況下對數據質量進行核對評估,對偏離方案的人群是否進入全分析集(Full analysis set,FAS)、符合方案集(Per protocol set,PPS)、安全性分析集(Safety set,SS)進行討論,以便確定統計分析計劃[3]。對盲態審核中方案偏離情況的分析和評價是研究結果科學可靠的必要保證。
為保證臨床試驗數據的真實性、可靠性和完整性,國家食品藥品監督管理總局(CFDA)于2015年7月22日發布了《關于開展藥物臨床試驗數據自查核查工作的公告》(以下簡稱“722公告”)[4],該公告規定已申報生產并且待審的新藥申請以及相關藥物臨床試驗項目均需申請人對臨床試驗數據進行自行檢查。這是我國臨床試驗數據核查理念變革的關鍵節點,代表著我國藥企、合同研究組織(Contract research organization,CRO)與藥物臨床試驗機構將積極推動臨床試驗的合規性。為了解我國臨床試驗管理中常出現的問題,進一步完善我國藥物臨床試驗法規與指導原則,本研究以某CRO公司的盲態數據審核表中臨床試驗項目的試驗分期、藥物種類、所處“722公告”前后時期作為分類標準,歸納項目所出現的偏離類型,分析偏離方案中的人群劃分情況。通過偏離類型、方案偏離的差異性及對試驗結果的影響分析,反映臨床試驗質量控制中存在的問題,提出改進措施,為藥物臨床試驗的質量管理提供參考,促進臨床試驗高質量地完成。
1 資料與方法
1.1 研究對象
收集廣州某CRO公司2010-2019年完成藥物臨床試驗項目的盲態數據審核表。由于Ⅰ期臨床試驗主要為藥動學(PK)或藥效學(PD)分析,偏離情況與其他分期差異較大,不易于合并分析,故本研究不納入Ⅰ期臨床試驗項目。
1.2 研究方法
1.2.1 收集試驗項目基本信息 收集項目的臨床試驗分期、藥物類型、試驗時間、病例數、中心數等信息。本研究中的不同臨床試驗分期包括Ⅱ、Ⅲ、Ⅳ和未分期臨床試驗,不同藥物類型包括中藥、化學藥及生物制品;另外以“722公告”為時間節點,將試驗項目分為“722公告”前結束的項目(以下簡稱“722公告”前)、“722公告”前啟動且“722公告”后完成的項目(以下簡稱跨越“722公告”)和“722公告”后啟動的項目(以下簡稱“722公告”后)三個階段。
1.2.2 分析項目方案偏離總體特征 按照盲態數據審核表的方案偏離情況進行劃分,定義超窗、違背納入與排除標準、脫落、合并用藥、未完成相應檢查、服藥依從性等不同方案偏離的類型。將參加試驗的受試者的偏離情況進行分析、歸類、量化、統計,將結果記錄于數據提取表中。方案偏離的類型定義見表1。
1.2.3 分析“722公告”前后的方案偏離情況 對“722公告”前后的方案偏離情況進行亞組分析,比較“722公告”前、跨越“722公告”和“722公告”后三個階段的項目所發生的方案偏離情況。
1.2.4 分析方案偏離對FAS人群劃分的影響 FAS是最大程度將入組的受試者都納入統計分析的數據集,對于已經被分配隨機號的受試者以最合理的方式最少地剔除,一般只有在發生重大方案違背、誤納、未服用過試驗藥物或隨機分配后無任何觀測數據的情況下才不納入FAS[5]。PPS是FAS中更加符合方案的病例。分析盲態數據審核表中方案偏離對FAS的影響以及方案偏離對納入FAS的人群是否納入PPS的影響,再結合方案偏離情況進一步分析對試驗療效結果的影響。
1.3 統計學方法
采用SPSS 20.0軟件對所得數據進行分析。計數資料以率表示;通過集中趨勢和離散趨勢進行正態分布檢驗,非正態分布的計量資料以中位數與四分位間距M(P25,P75)表示。不同試驗分期和試驗時間所發生方案偏離的差異用多個樣本比較的Kruskal-Wallis檢驗分析,不同藥物類型發生方案偏離的差異用兩樣本比較的Mann-Whitney檢驗分析。P<0.05表示差異有統計學意義。
2 結果
2.1 納入項目的基本情況
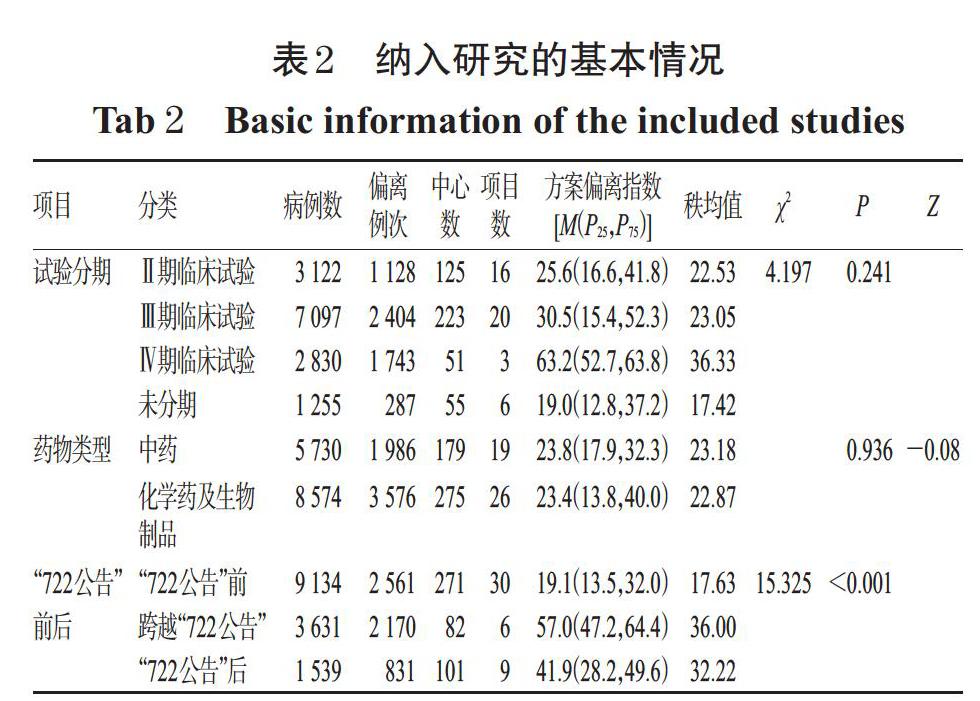
通過歸類分析,最終納入45個符合條件的臨床試驗項目,共涉及454個中心、14 304例病例及5 562例次方案偏離。納入研究的基本情況見表2[表中方案偏離指數代表每100個病例發生偏離的例次數,方案偏離指數=100×(偏離病例次數/病例數)。
由表2可知,分別對不同臨床試驗分期、不同藥物類型、“722公告”前后的偏離方案的差異性進行分析,結果顯示,不同試驗分期和不同藥物類型的臨床試驗項目發生方案偏離的程度沒有顯著性差異(P>0.05);“722公告”前、跨越“722公告”和“722公告”后臨床試驗項目的方案偏離指數的差異有統計學意義(χ2=15.325,P<0.001),可認為三個階段方案偏離的程度顯著不同。
2.2 對盲態審核中臨床試驗方案偏離類型的總體分析
方案偏離常見類型的分布情況見表3。
由表3可知,臨床試驗中最常見的方案偏離類型依次是超窗、違背納入與排除標準、脫落,分別占總體偏離情況的36.88%、20.71%、18.43%。
2.3 “722公告”前后偏離方案的亞組分析
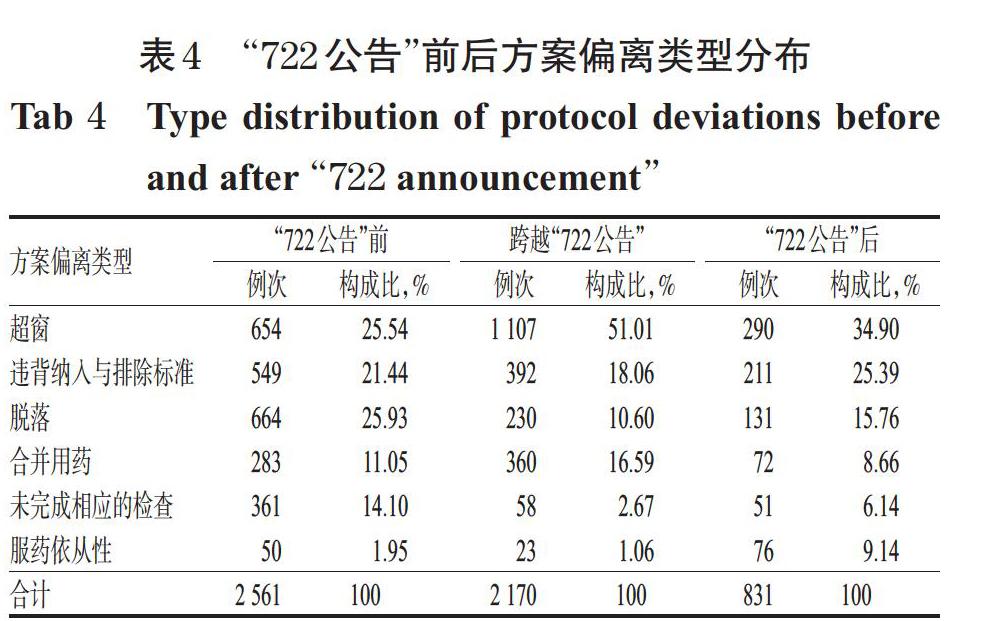
以所處“722公告”前后時期為分類標準,對比“722公告”前、跨越“722公告”以及“722公告”后三個階段方案偏離的分布情況,結果見表4。
由表4可知,“722公告”前后藥物臨床試驗方案偏離情況的分布存在差異:跨越“722公告”期間的臨床試驗項目出現超窗偏離的比例高達51.01%,“722公告”后期開展的臨床試驗項目出現超窗、違背納入與排除標準和服藥依從性等偏離的比例均大于“722公告”前結束的試驗項目所出現的情況。
2.4 方案偏離對FAS人群劃分的影響
本研究入選的項目中可納入FAS的方案偏離有2 507例次,FAS中未進入PPS的方案偏離例次占53.99%,其中脫落、合并用藥和服藥依從性偏離例次分別占19.51%、4.29%和2.00%。而進入與未進入PPS的超窗和違背納入與排除標準的方案偏離例次比例相當。方案偏離中納入FAS中的PPS情況如表5所示。
3 討論與建議
3.1 方案偏離類型的分析
超窗是臨床試驗中最常出現的問題,訪視時間的延后或提前會引起高估或低估藥物的療效。根據超窗對試驗結果的影響程度可分為嚴重超窗與輕度超窗兩種類型:嚴重超窗是指受試者關鍵檢查或訪視日期遠超出規定的時間窗口,會明確影響試驗藥物的有效性評價,如果涉及到主要療效指標嚴重超窗的情況,最后會從FAS集中剔除;輕度超窗是時間窗在可容忍的范圍內,依從性符合要求或非關鍵檢查超窗,不影響有效性評價。超窗通常由受試者或者研究者依從性不佳引起[6],如受試者對試驗不配合、態度不重視或研究者對試驗方案設計不恰當、不熟悉。本研究出現的超窗多由研究者間接造成,因此試驗前應對研究者進行全方位的培訓,增強研究者的GCP意識和對方案的熟悉程度,研究人員在訪視前也需提前通知受試者。
違背納入與排除標準也是臨床試驗中常出現的問題,多為研究人員對方案執行意識的薄弱導致誤納入,以及由于納入受試者病情較為復雜,在試驗期間發生不可控的生理或病理變化引起。遵守納入與排除標準、篩選合格的受試者是藥物臨床試驗順利開展的前提條件。篩選受試者時應去除病情過于復雜、可能影響療效評價、不適合參與試驗的人群,一般包括妊娠婦女、肝腎功能不全、對試驗藥物可能過敏或合并其他重大疾病等人群[7-8]。方案執行前,研究者需對方案的科學性以及可操作性進行評估。在對臨床監查員(CRA)和臨床協調員(CRC)進行培訓時,應重點強調納入與排除標準,指出易違背的關鍵點,要求其嚴格按照方案執行,研究者及CRC需對相關結果認真核對,減少病例誤納入的可能性。在試驗啟動會上,還應向研究者強調試驗操作應以方案要求為準,對試驗過程中的疑問需及時記錄并反饋給CRA或者項目組成員。
脫落為臨床試驗中較易發生的問題之一,可能會引起臨床試驗的療效指標和安全指標缺失,產生結果偏倚。病例脫落與受試者的依從性存在較大的相關性。受試者常因外出打工、交通不方便、換工作、搬家、家人反對或懷疑試驗藥物的效果而要求退出或者失訪。受試者的年齡、性別、受教育程度、性格、成長環境、病情程度不同,其對臨床試驗的依從性也會有所差異。研究者在篩選受試者時應多方面評估受試者的依從性,優先選擇依從性良好的受試者;試驗方案的設計方面,盡可能減少訪視點,簡化試驗流程,增加方案的可操作性[9]。CRA訪視時要關注受試者的用藥情況,通過查看病歷報告、溯源醫院信息系統、門診開藥信息,來確認合并治療情況;一旦發現受試者合并使用方案禁止的其他藥物,須及時記錄,并報告方案偏離。
本研究表明,大部分偏離原因與質量保證和質量控制有關。在藥物臨床試驗中,應加強第三方稽查、研究者監督、項目組成員質控及CRA的監查等,發生質量問題后及時采取糾正措施;對于多中心發現的問題應進行匯總分析,若項目存在共性問題,可對同類問題采取預防措施,降低同類型問題再次發生的概率。
3.2 方案偏離的差異性分析
“722公告”后,CFDA不斷完善臨床試驗相關法律法規,頒布臨床試驗數據管理有關的指導規范與指南。在借鑒國際規范、技術指南并結合我國現狀的基礎上,CFDA于2016年發布《臨床試驗數據管理工作技術指南》《藥物臨床試驗的生物統計學指導原則》《藥物臨床試驗數據管理與統計分析的計劃和報告指導原則》《臨床試驗的電子數據采集技術指導原則》,以確保藥物臨床試驗數據的規范性、完整性和真實性。2017年6月1日,CFDA成為國際人用藥品注冊技術協調會(International Council for Harmonization,ICH)成員,加入ICH是國際對我國藥品監管水平和能力認可的標志,也意味著對我國臨床試驗的質量提出了更高的要求[10]。
本研究結果顯示,“722公告”后,超窗、違背納入與排除標準及服藥依從性偏離情況的發生率升高,但這一趨勢不代表“722公告”后開展的臨床試驗項目實際發生的偏離率較之前高。“722公告”核查公告的原則為自查糾錯從寬、被查處理從嚴、嚴懲故意造假、允許規范補正,專家組成員需在此原則下開展現場檢查工作,確保臨床試驗數據的真實、完整與可靠性,確保受試者的安全和權益受到保護。面對有史以來最嚴的數據核查要求,大部分項目被撤回或整改,以往被忽略的違背方案或者漏報的試驗項目紛紛上報。跨越“722公告”階段的項目正好面臨整改階段,方案偏離被發現并且上報的數量要高于“722公告”前開展的項目。可認為“722公告”政策出臺后,研究者主動報告、自查等因素致使被發現并且上報的方案偏離更接近實際情況,發布藥物臨床試驗數據核查公告后相關人員的GCP意識有所提升,對質量的控制更加規范,更加關注數據的真實性和完整性。
3.3 方案偏離對臨床試驗結果的影響
臨床試驗包括優效性、等效性和非劣效性臨床試驗,FAS和PPS常用于試驗藥物的療效性分析,兩者在不同的試驗中所起的作用不同[11]。由于FAS的分析結果比較保守,依從性差的受試者的加入可能導致藥物療效被低估,在優效性試驗中宜采用FAS作為主要分析集。PPS可顯示試驗藥物按規定使用的效果,相對于上市后的藥物療效,可能高估了實際療效。在對等效性或非劣效性臨床試驗的再統計分析時,用FAS所得到的結果不一定保守,此時可以結合PPS的分析結果。
[ 4 ] 國家食品藥品監督管理總局.關于開展藥物臨床試驗數據自查核查工作的公告[EB/OL]. (2015-07-22) [2020- 03-02]. http://www.nmpa.gov.cn/WS04/CL2182/299803.html.
[ 5 ] 國家食品藥品監督管理總局.藥物臨床試驗的生物統計學指導原則[S]. 2016-06-01.
[ 6 ] 王曉霞,李育民,高瑾,等.藥物臨床試驗中受試者的依從性問題研究[J].中國藥物與臨床,2009,9(6):507-508.
[ 7 ] 張強,蒙萍,單愛蓮.關于藥物臨床試驗方案中納入、排除標準的若干思考[J].中國臨床藥理學雜志,2017,33(2):99-101.
[ 8 ] 李雪迎.臨床實驗設計三要素之研究對象[J].中國介入心臟病學雜志,2014,22(5):317.
[ 9 ] 陳國霞.淺談新藥臨床試驗中受試者依從性的管理[J]. 中國民族醫藥雜志,2016,22(3):55-56.
[10] 郭薇,謝林利,曹麗亞,等.加入ICH對我國藥物臨床試驗機構工作的影響和思考[J].中國藥房,2019,30(11):1445-1448.
[11] 李雪迎.臨床試驗研究統計學設計方法簡述-優效性設計、等效性設計以及非劣效性設計[J].中國介入心臟病學雜志,2014,22(8):482.
[12] LESAFFRE E,DE K. Estimating the power of comp-? ? ?liance-improving methods[J]. Control Clin Trials,2000,21(6):540-551.
[13] URQUHART DJ. Role of patient compliance in clinical pharmacokinetics[J]. Clin Pharmacokinet,1994,27(3):202-215.
[14] 張抗,李文元,馮碩,等.臨床試驗中脫落、退出和失訪病例的統計學處理和報告規范[J].中醫雜志,2016,57(14):1204-1207.
[15] CRUTZEN R,VIECHTBAUER W,KOTZ D,et al. No differential attrition was found in randomized controlled trials published in general medical journals:a meta-analysis[J]. J Clin Epidemiol,2013,66(9):948-954.
[16] VAN DLM,LORENZ TJ. Addressing missing data in cli- nical trials[J]. Ann Intern Med,2011,154(2):113-117.
[17] 衡明莉,陳麗嫦,王駿.臨床試驗中缺失數據處理方法研究[J].中國臨床藥理學雜志,2019,35(22):2948-2952.
[18] 張念先.臨床試驗常用缺失數據處理方法的局限性分析[J].中國新藥與臨床雜志,2009,28(9):659-662.
(收稿日期:2020-04-08 修回日期:2020-07-03)
(編輯:劉明偉)
猜你喜歡
知音勵志·社科版(2016年9期)2016-11-09 08:21:16
中國科技博覽(2016年22期)2016-11-01 15:06:27
中國科技博覽(2016年22期)2016-11-01 14:20:50
中國科技博覽(2016年22期)2016-11-01 12:54:47
現代企業文化·理論版(2016年14期)2016-10-21 10:29:30
中國科技博覽(2016年19期)2016-10-19 13:44:57
中國科技博覽(2016年19期)2016-10-19 12:36:32
中國市場(2016年36期)2016-10-19 04:22:24
科學與財富(2016年28期)2016-10-14 22:54:28
科學與財富(2016年28期)2016-10-14 22:41:32
