一種選擇性檢測肼的近紅外熒光增強型探針的合成
2020-09-21 11:59:18崔桂玲胡曰富齊慶蓉
合成化學 2020年9期
關鍵詞:檢測
崔桂玲, 宋 艷, 胡曰富, 齊慶蓉
(四川大學 華西藥學院,四川 成都 610041)
肼作為一種化學物質,由于其堿性和還原性,使其在醫藥、化學、催化、農業等領域有廣泛應用,同時,肼也是一種高能量原料,所以也被應用于航天領域,然而該物質不僅有毒,還有致癌作用,微小量的泄露不僅會造成環境污染,也會對人體的肝、腎、肺、中樞神經、呼吸系統等造成傷害[1-5],因此對于微量肼的檢測至關重要。
傳統的檢測肼的分析方法包括電化學分析法、色譜法和分光光度法,但這些分析方法不僅復雜耗時,而且靈敏度低[6-9]。相比于傳統的肼檢測方法,熒光分析法具有高靈敏性、高選擇性、操作簡便、成本低等優點[10-12],因此更受歡迎。文獻報道的主要檢測肼的基團有乙酰基、4-溴丁酸酯、乙酰丙酸鹽、鄰苯二甲酰亞胺、醛、丙二腈等,
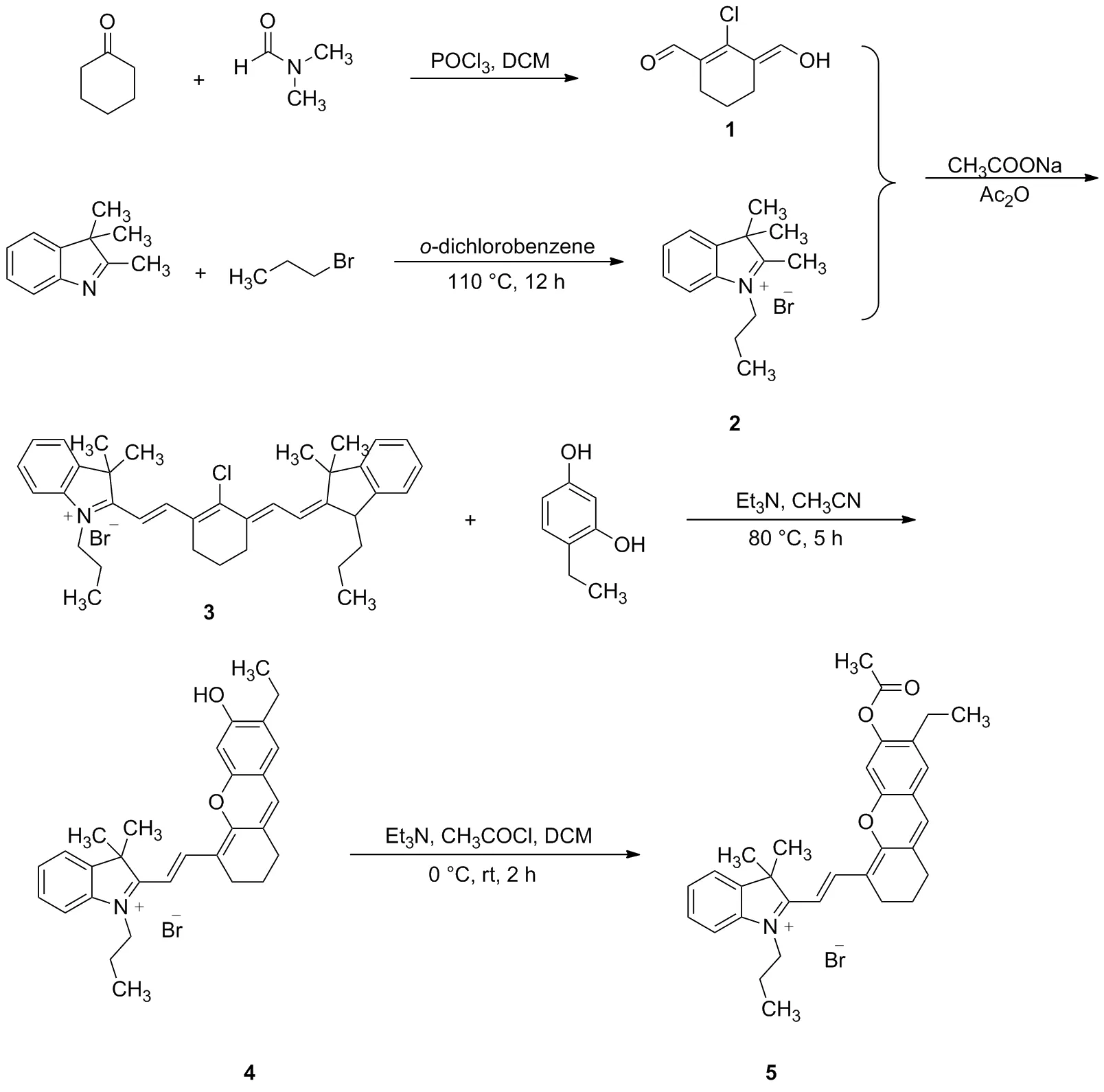
Scheme 1
但大部分探針的最大紫外吸收和發射均位于紫外可見光區,不適合生物成像。因近紅外區的光具有較低的背景熒光,較低的光毒性和較深的穿透性[13-14],所以近紅外熒光探針更具有應用價值。
2016年,Zhang等[13]發現了用乙酸酯作為識別位點的檢測肼的近紅外探針,2018年,Lu等[15]報道了用溴丁酸酯作為識別基團的檢測肼的近紅外探針,這類探針通過一個可以調控的羥基使探針及檢測肼后產物的最大發射波長均位于近紅外區,具有明顯的降低自熒光、低毒性、深度滲透等優點,但兩者最大發射波長很接近,難以觀察到探針和產物各自熒光變化的線性。2012年,Lin等[16]報道在半花菁類分子(Chart 1)羥基的鄰位引入氯原子可以改變半花菁分子的部分光學性質,但類似報道很少。鑒于此本文設計并合成了一個新型的檢測肼的熒光探針,并對其進行了光譜分析,研究其對肼的選擇性。以2,3,3-三甲基-3H-吲哚與正丙基溴為原料反應合成2,3,3-三甲基-1-丙基-3H-吲哚鹽2,繼而與2-氯-3-羥次甲基環己烯醛縮合生成2- (2-(2-氯-3-((3,3-二甲基-1-丙基-1,3-二氫-2H-吲哚-2-亞烷基)亞乙基)-1-環己烯-1-)乙烯基)-3,3-二甲基-1-丙基-3H-吲哚鹽3,再與4-乙基間苯二酚在三乙胺催化下反應生成2-(2-(7-乙基-6-羥基-2,3-二氫-1H-呫噸-4-)乙烯基)-3,3-二甲基-1-丙基-3H-吲哚鹽4,最后與乙酰氯經成酯反應生成半菁類探針2-(2-(7-乙基-6-乙酰氧基-2,3-二氫-1H-呫噸-4-)乙烯基)-3,3-二甲基-1-丙基-3H-吲哚鹽5(Chart 2),探針結構經過1H NMR、13C NMR和HR-MS(ESI)表征。
1 實驗部分
1.1 儀器與試劑
UV-2450型紫外可見分光光度計;Varian-Unity 400 MHz型核磁共振儀;Waters Q-TOF-Premier型質譜儀;Varian Cary Eclipse Fluoromete型熒光光譜儀。
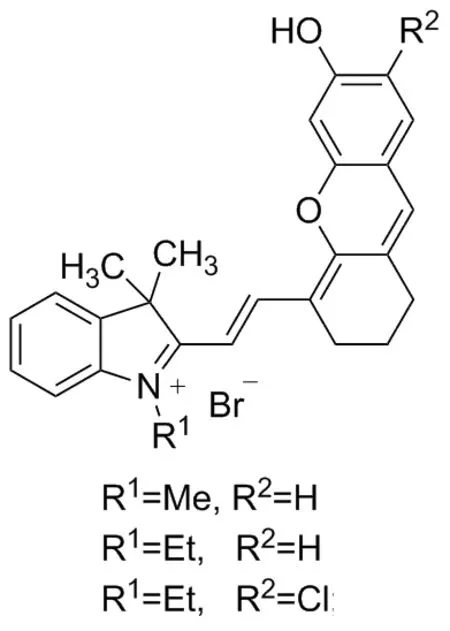
Chart 1
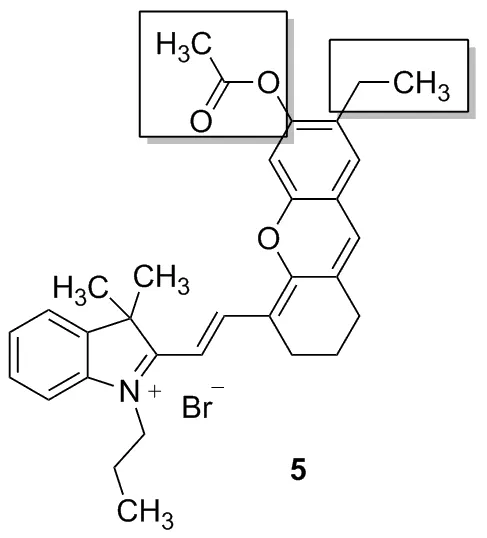
Chart 2
其余所用試劑均為分析純。
1.2 合成
(1)1的合成[17-18]
在干燥的500 mL三頸瓶中加入N,N-二甲基甲酰胺37.2 g(0.5 mol)和二氯甲烷50 mL, 0 ℃下依次滴加三氯氧磷62.5 g(0.4 mol),環己酮10.0 g(0.1 mol), 80 ℃回流4 h;減壓蒸除溶劑,冷卻至室溫后傾至冰水中,析出大量黃色固體,抽濾,濾餅用冰水洗滌3次,真空干燥,丙酮重結晶后得黃色固體19.0 g,收率51%, m.p.127~128 ℃;1H NMR(DMSO-d6, 400 MHz)δ: 10.84(s, 1H, CHO), 2.53~2.47(m, 1H, CH), 2.36(t,J=6.1 Hz, 4H, CH2), 1.58(p,J=6.3 Hz, 2H, CH2)。
(2)2的合成[17-19]
在干燥的250 mL三頸瓶中加入2,3,3-三甲基-3H-吲哚5.0 g(31.5 mmol)、正丙基溴4.7 g(38.1 mmol)和鄰二氯苯15 mL,110 ℃攪拌12 h(TLC檢測)。冷卻至室溫,傾倒入100 mL異丙醚中,大量粘性固體析出,靜置,傾倒出上清液,加少量丙酮并攪拌,大量固體析出,抽濾,濾餅用異丙醚洗滌三次,得棕紅色固體產物24.5 g。該產品不經純化直接用于下一步。
(3)3的合成[17-18]
在干燥的100 mL三頸瓶中加入24.5 g(6.0 mmol)、11.9 g(11.0 mmol)、乙酸鈉1.8 g(22.2 mmol)和乙酸酐8 mL,于室溫攪拌2 h,(TLC檢測)。加100 mL異丙醚,有大量綠色固體析出,抽濾,濾餅用異丙醚洗滌,得綠色固體產物36.9 g。該產品不經純化直接用于下一步。
(4)4的合成[17]
在干燥的100 mL三頸瓶中加入3300.0 mg(49.6 mmol)、 4-乙基間苯二酚100.0 mg (72.4 mmol)、三乙胺270.0 mg(2668.2 mmol)和乙腈8 mL,回流5 h(TLC檢測)。冷卻至室溫,減壓蒸除溶劑,殘余物經硅膠柱層析(洗脫劑:二氯甲烷/甲醇=100/1~30/1,V/V)純化得青綠色固體4170.0 mg,收率68%;1H NMR(Methanol-d4, 400 MHz)δ: 8.15(d,J=13.4 Hz, 1H, CH=CH), 7.56(s, 1H, ArH), 7.30(d,J=7.4 Hz, 1H, ArH), 7.20(t,J=7.7 Hz, 1H, ArH), 7.17(s, 1H, ArH), 6.99(t,J=7.3 Hz, 1H, ArH), 6.95(d,J=8.0 Hz, 1H, ArH), 6.41(s, 1H, CH), 5.76(d,J=13.5 Hz, 1H, CH=CH), 3.82(t,J=7.4 Hz, 2H, CH2), 2.66(t,J=6.1 Hz, 2H, CH2), 2.57(t,J=6.7 Hz, 2H, CH2), 2.52(t,J=7.5 Hz, 2H, CH2), 1.82(p,J=6.3 Hz, 2H, CH2), 1.72(p,J=7.4 Hz, 2H, CH2), 1.61(s, 6H, CH3), 1.13(t,J=7.5 Hz, 3H, CH3), 0.94(t,J=7.4 Hz, 3H, CH3); HR-MS(ESI)m/z: Calcd for C30H34NO2Br{[M-Br]+}440.2584, found 440.2578。
(5)5的合成[15-16]
在干燥的25 mL單頸瓶中加入421.9 mg(42.2 nmol)、三乙胺60.0 mg(59.3 nmol)和二氯甲烷25 mL,于冰鹽浴中攪拌,緩慢滴加乙酰氯110.0 mg(1.4 mmol)的二氯甲烷溶液2 mL,滴畢,室溫攪拌2 h(TLC檢測)。減壓蒸除溶劑,殘余物經硅膠柱層析(洗脫劑:二氯甲烷/甲醇=100/1~30/1,V/V)純化得青綠色固體5150.0 mg,收率93%;1H NMR(CDCl3, 400 MHz)δ: 8.61(d,J=14.9 Hz, 1H, CH=CH), 7.55~7.37 (m, 4H, ArH), 7.27(s, 1H, ArH), 7.15(s, 1H, ArH), 7.04(s, 1H, CH), 6.70(d,J=14.8 Hz, 1H, CH=CH), 4.58(t,J=7.1 Hz, 2H, CH2), 2.80(t,J=6.1 Hz, 2H, CH2), 2.74(t,J=6.1 Hz, 2H, CH2), 2.58(q,J=7.5 Hz, 2H, CH2), 2.39(s, 3H, CH3), 2.08~1.87(m, 4H, CH2), 1.79(s, 6H, CH3), 1.21(t,J=7.5 Hz, 3H, CH3), 1.08(t,J=7.3 Hz, 3H, CH3);13C NMR(CDCl3, 100 MHz)δ: 178.30, 169.14, 160.27, 151.26, 150.81, 146.14, 142.00, 141.45, 133.86, 131.58, 130.17, 129.34, 127.76, 127.56, 122.52, 119.96, 115.40, 113.36, 109.96, 105.98, 50.91, 47.81, 29.49, 28.24, 24.56, 22.80, 21.59, 21.00, 20.26, 13.97, 11.59; HR-MS(ESI)m/z: Calcd for C32H36NO3Br{[M-Br]+}482.2690, found 482.2693。
2 結果與討論
2.1 紫外-可見光譜
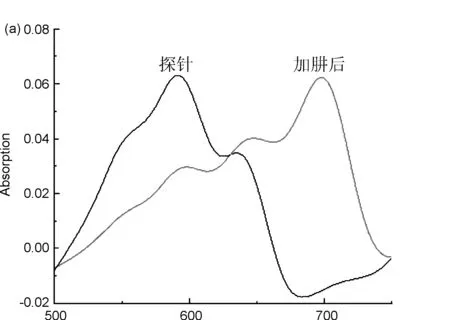
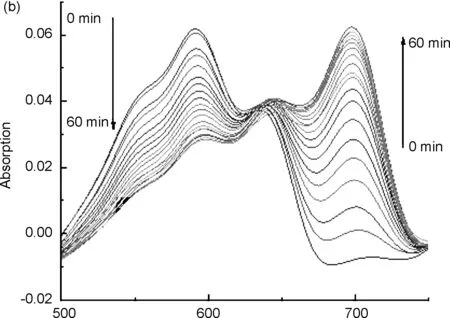
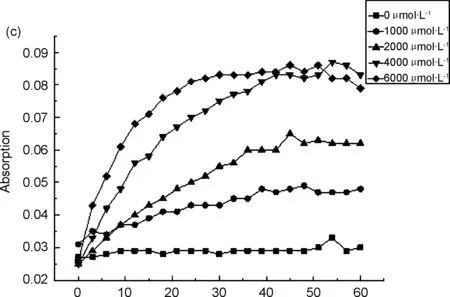
用15% DMSO PBS溶液(pH=7.4)分別配制20 μmol/L的探針5標準溶液和2000 μmol/L的肼溶液,兩者等體積混合后,在避光條件下,每三分鐘取樣進行紫外掃描。由圖1a可知,探針5的最大紫外吸收波長為598 nm,與肼反應后生成的產物最大紫外吸收波長為702 nm;由圖1b可知,探針與肼反應60 min內,探針最大紫外吸收值逐漸降低,產物最大紫外吸收值逐漸升高,隨著反應時間的延長,598 nm處的最大紫外吸收值逐漸降低,702 nm處的最大紫外吸收值逐漸升高,且加肼前后混合液的顏色變化明顯,加肼之前為藍色,加肼反應之后為青綠色,表明探針5能夠檢測肼;由圖1c可以看出,探針與肼的反應時間隨著肼濃度的增大而加快。與肼反應之前,探針5的羥基是以乙酸酯的形式存在,羥基氧原子給電子能力降低,最大紫外吸收波長較低,當與肼反應之后,產物主要以氧負離子的形式存在,氧原子給電子能力增強,故最大紫外吸收波長紅移[16]。
2.2 熒光發射光譜
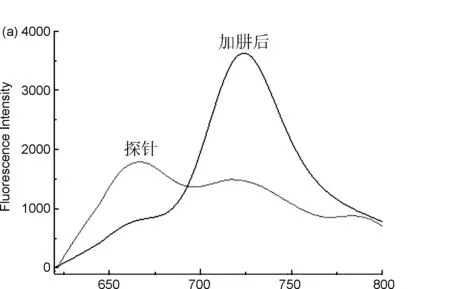
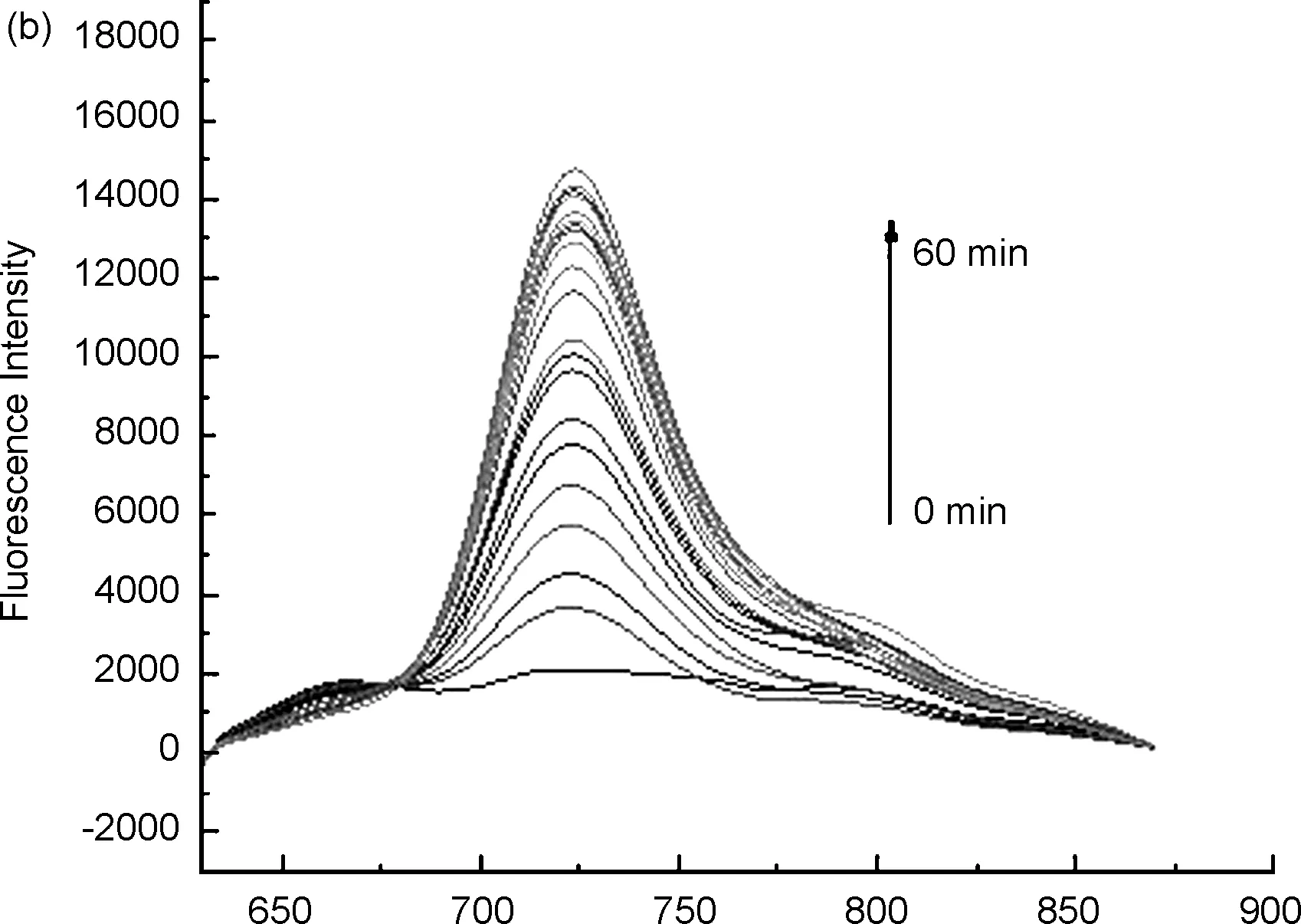
用DMSO配制濃度為640 μmol/L探針標準溶液,用15%DMSO的PBS緩沖溶液(pH=7.4) 將探針5標準溶液稀釋成20 μmol/L,分別與不同濃度的肼等體積混合反應,得到熒光發射光譜(圖2c)。用598 nm進行激發,可得到探針5的最大發射波長為666 nm,與肼反應之后,產物最大發射波長位于723 nm處(圖2a),且產物的熒光強度隨著時間的延長大幅度增加,探針的熒光強度緩慢減小(圖2b)。
λ/nm
λ/nm
t/min
λ/nm
λ/nm
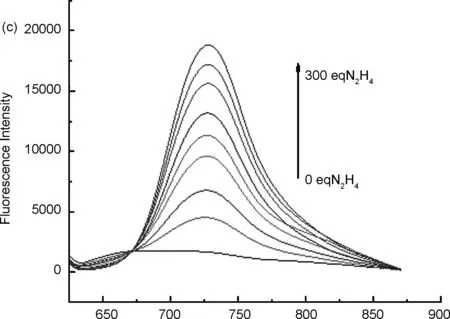
λ/nm圖2探針5的熒光發射光譜(a: 加肼前后的變化情況; b: 探針與不同濃度肼混合后在666 nm和723 nm處的熒光強度變化情況; c: 探針對不同濃度肼的熒光強度變化)
根據光誘導電子轉移原理(PET),當探針5的識別基團乙酰基處于自由態時,其HOMO軌道上的電子可以向熒光團的HOMO轉移,致使熒光團被激發到LOMO軌道上的激發態電子不能返回基態產生熒光或熒光很弱,而乙酰基與肼結合之后,乙酰基的HOMO上的電子無法轉移到熒光團的HOMO軌道上,使PET過程無法進行,這時熒光團的激發態電子可以返回基態產生熒光,故產物熒光遠強于探針[20]。
2.3 選擇性評價
選擇性是評價探針的重要指標之一,因此本實驗研究了探針5對可能的常見干擾物質 (2-羥基乙基胺、 KCl、 Na2SO4、 CaCl2、 MgSO4、 CuSO4、 ZnSO4、 FeCl3、 FeSO4、 NaBr、 CH3COONa、 K2CO3、 NaH2PO4、 NH4Cl、 NH3.H2O、二乙胺、尿素、硫脲、L-甘氨酸、L-賴氨酸、L-精氨酸、異煙肼和乙酰肼)的響應情況,分別向濃度為20 μmol/L的探針標準溶液中加入等體積的2000 μmol/L的待測物質溶液,室溫避光放置48 min后,依次取樣進行熒光掃描,結果表明,該探針5對上述物質無明顯響應,對肼具有良好的選擇性。
本文設計合成了一個可用于檢測肼的近紅外小分子熒光探針,通過可調控基團羥基,使探針5在與肼反應前后共軛結構發生改變,從而引起熒光強度的變化。探針5最大紫外吸收波長為598 nm,最大熒光發射波長為666 nm,與肼反應后的產物最大紫外吸收波長為702 nm,最大熒光發射波長為723 nm,當肼的濃度在0~1000 μmol/L時,熒光強度與濃度具有良好的線性關系,相關系數R2=0.9919,檢測限為2.93 μmol/L。同時,該探針的最大發射波長位于近紅外,有利于克服生物體自身熒光的干擾,有望成為一種理想的檢測肼的熒光探針。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
