超高效液相色譜-串聯質譜法同時測定黃芩提取物中4種黃酮類成分
2020-08-06 06:28:54
理化檢驗-化學分冊 2020年7期
(烏蘭察布醫學高等專科學校,烏蘭察布 012000)
黃芩又名山茶根、土金茶根,為唇形科黃芩屬多年生草本植物,在我國北方各省均有種植。黃芩提取物為黃芩干燥根經加工制成的淡黃色粉末,具有清熱燥濕、瀉火解毒、安胎、止血等功效[1-4],臨床上用于治療上呼吸道感染、肺熱咳嗽、肝火頭痛、目赤腫痛、濕熱黃疸、胎動不安、高血壓、癰腫癤瘡等癥[5-6],其具有藥理活性的化學成分主要為黃芩素、黃芩苷、褪黑素、5-羥色胺、漢黃芩苷、野黃芩苷等[7-9]。
目前黃芩的評價主要是通過其藥理活性化學成分測定或者色譜指紋圖譜等進行品種鑒別、產地來源分析,測定方法包括薄層色譜法[10]、液相色譜法[2,9,11-12]和液相色譜-質譜法(LC-MS/MS)[8,13-14],而中國藥典(2015年版)中黃芩提取物定量指標成分僅有黃芩苷一項,不能完全反映黃芩提取物的整體質量。對不同來源的黃芩提取物進行有效成分的定性定量鑒別已成為規范黃芩提取物的質量控制并保證其臨床用藥安全性、有效性的重要途徑。
本工作針對7份不同產地的黃芩提取物,采用甲醇超聲提取,經ACQUITY UPLC HSS T3色譜柱分離,質譜檢測器測定,建立了超高效液相色譜-串聯質譜法(UHPLC-MS/MS)測定黃芩提取物中黃芩素、黃芩苷、漢黃芩苷、野黃芩苷等4種黃酮類成分的分析方法,旨在為黃芩提取物的質量控制及其藥理活性化學成分研究提供測定方法。
1 試驗部分
1.1 儀器與試劑
Waters ACQUITY UPLC/Xevo TQ MS型超高效液相色譜-串聯質譜儀;Milli-Q 型超純水器;JP-060型超聲波清洗機;5415D 型高速冷凍離心機;Syncore Polyvap型多樣品平行蒸發儀。
4種黃酮類成分的標準儲備溶液:1 000 mg·L-1,稱取黃芩素、黃芩苷、漢黃芩苷、野黃芩苷的標準物質各50.0 mg,分別置于50 mL容量瓶中,用適量甲醇溶解,并用甲醇定容至50.0 mL。
黃芩素、黃芩苷、漢黃芩苷、野黃芩苷標準物質均為色譜純;其余試劑均為分析純;試驗用水為超純水。
1.2 儀器工作條件
1)色譜條件 ACQUITY UPLC HSS T3 色譜柱(2.1 mm×150 mm,1.8μm),柱溫35 ℃;流量0.2 mL·min-1;進樣體積10μL。流動相:A 為0.01%(質量分數,下同)氨水,B 為乙腈。梯度洗脫程序:0~4.0 min 時,A 由80%降至20%;4.0~6.0 min時,A 由20%降至5%,保持2.5 min;8.5~12.0 min時,A 由5%升至80%,保持3 min。
2)質譜條件 采用電噴霧離子源(ESI),負離子模式;離子源溫度150 ℃,脫溶劑溫度350 ℃;毛細管電壓3.5 kV;碰撞氣為氬氣,流量0.15 mL·min-1;錐孔氣為氮氣,流量50 L·h-1;脫溶劑氣為氮氣,流量600 L·h-1;多反應監測(MRM)模式。其余質譜參數見表1,其中,“*”為定量離子。
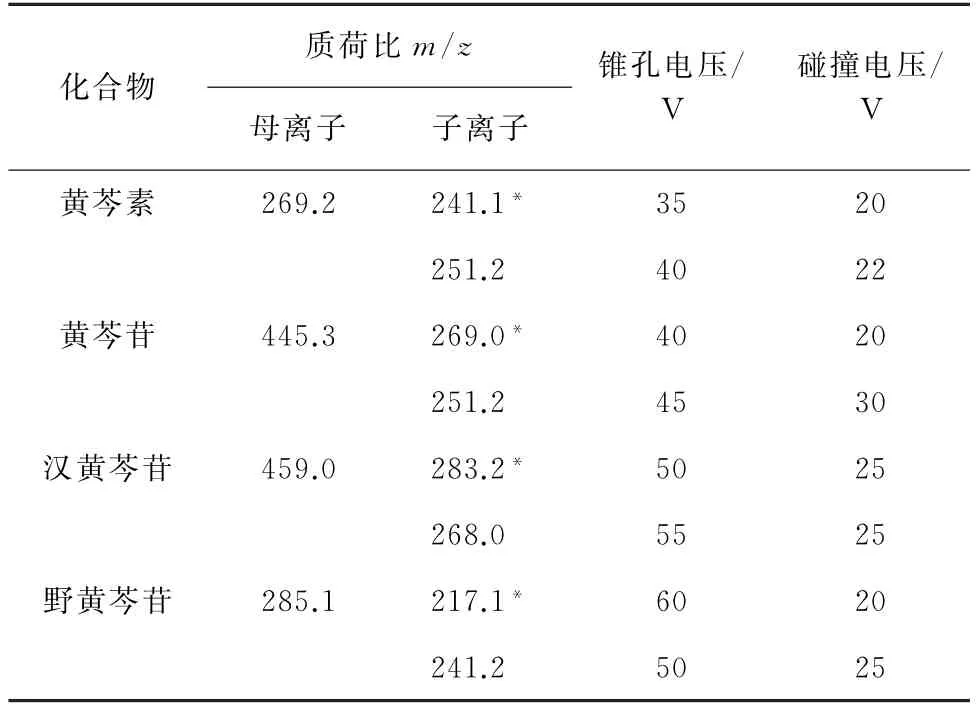
表1 質譜參數Tab.1 MS parameters
1.3 試驗方法
7份不同產地(山東沂蒙山區、甘肅慶陽、內蒙古赤峰、吉林延邊、河北承德、山西臨汾、陜西渭南)的黃芩提取物,購自某公司。
稱取黃芩提取物粉末樣品約0.1 g置于50 mL離心管中,加入4 mL水,渦旋30 s,再加入6 mL甲醇,渦旋混勻后超聲提取(超聲功率300 W,超聲頻率28 kHz)30 min,以12 000 r·min-1轉速離心10 min,收集上清液。殘渣加入4 mL水,渦旋30 s,再加入6 mL甲醇,渦旋混勻后超聲提取(超聲功率300 W,超聲頻率28 kHz)30 min,以12 000 r·min-1轉速離心10 min,合并上清液,用水定容至50.0 mL,過0.22μm 微孔濾膜后按儀器工作條件進行測定。
2 結果與討論
2.1 色譜-質譜行為
按儀器工作條件對4種黃酮類成分的混合標準溶液進行測定,色譜圖見圖1。
4種黃酮類成分的二級質譜圖見圖2。
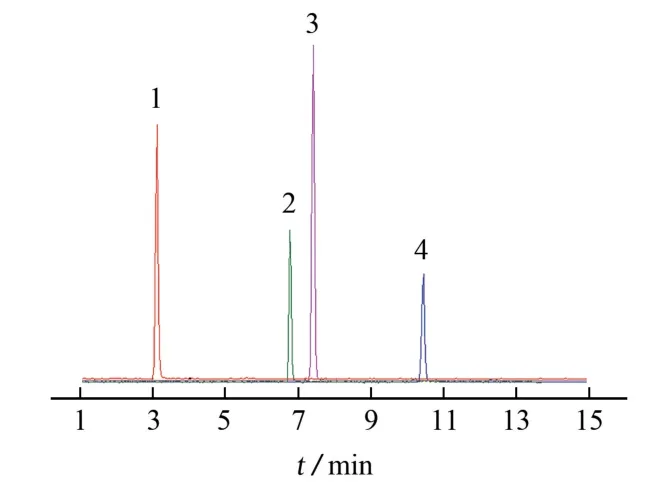
圖1 4種黃酮類成分混合標準溶液的總離子流色譜圖Fig.1 TIC chromatogram of mixed standard solution of 4 flavonoids
2.2 質譜條件的選擇
采用蠕動泵將1.0 mg·L-1的4種黃酮類成分混合標準溶液以1.0μL·min-1流量連續注入ESI中,分別采用正離子模式和負離子模式對其進行一級質譜分析,得到準分子離子峰(母離子),再對各準分子離子峰進行二級質譜分析,得到碎片離子峰(子離子)。結果表明:4種黃酮類成分在正離子模式和負離子模式下均有響應,但在負離子模式下產生的[M-H]-峰質譜信號強度較強,可能是由于這4種黃酮類成分結構中的羥基在負離子模式下易失去H 而形成穩定的[M-H]-峰。采用二級質譜掃描模式對準分子離子[M-H]-峰進行子離子掃描,優化碰撞電壓和錐孔電壓,確定各化合物的二級質譜圖,并使各碎片離子的質譜信號強度達到最高,選取豐度較大的兩個碎片離子作為定性離子和定量離子。試驗選擇的質譜條件見1.2節。
2.3 色譜柱的選擇
試驗考察了ACQUITY UPLC HSS T3 色譜柱、UPLC BEH C18色譜柱、UPLC HSS C18色譜柱、UPLC HSS C18SB 色譜柱、UPLC BEH HILIC 色譜柱等5種超高效液相色譜柱對黃芩提取物中4種黃酮類成分分離情況的影響。結果表明:當采用ACQUITY UPLC HSS T3 色譜柱時,4 種黃酮類成分完全分離,保留時間(3.16~10.47 min)適中,且各化合物的色譜峰峰形尖銳對稱。試驗選用ACQUITY UPLC HSS T3色譜柱。
2.4 流動相的選擇

圖2 4種黃酮類成分的二級質譜圖Fig.2 MS2 spectra of 4 flavonoids
試驗考察了甲醇、乙腈、水、0.01%(體積分數,下同)甲酸溶液、0.1%甲酸溶液、0.01%氨水、0.1%氨水等組合為不同流動相體系時對黃芩提取物中4種黃酮類成分的色譜分離效果和質譜離子化效率的影響。結果表明:乙腈-水體系作為流動相時,4種黃酮類成分的質譜響應優于甲醇-水體系作為流動相時的質譜響應;當流動相體系的pH 小于7時,在負離子模式下易產生[M+H]+峰和[M+Na]+峰,其中黃芩素離子化效率和質譜信號強度較高,其他3種化合物的離子化效率和質譜信號強度不高;當流動相體系的pH 大于7時,在負離子模式下易產生[M-H]-峰,尤其是采用0.01%氨水-乙腈體系作為流動相時,經梯度洗脫,4種黃酮類成分得到了良好的色譜分離效果和質譜信號強度,色譜圖中基線平穩,峰寬較窄,峰形尖銳對稱。試驗選擇流動相為0.01%氨水-乙腈體系。
2.5 提取溶劑的選擇
水分含量較低的樣品在進行分析時,通常在樣品前處理時加入一定量水,以提高后續的提取效率。試驗考察了水的加入量對黃芩提取物中4種黃酮類成分提取效果的影響。結果表明:水加入量不大于4 mL時,4種黃酮類成分的提取率隨水加入量的增大而增大;水加入量大于4 mL 時,4種黃酮類成分的提取率呈下降趨勢。試驗選擇水加入量為4 mL。
試驗考察了提取溶劑依次為甲醇、乙醇、乙腈、丙酮、異丙醇、乙酸乙酯、二氯甲烷時對黃芩提取物中4種黃酮類成分提取效果的影響。結果表明:4種黃酮類成分可溶于甲醇、乙醇、乙腈和丙酮,微溶于異丙醇、乙酸乙酯和二氯甲烷;4種黃酮類成分的提取率由大到小對應的提取溶劑依次為乙腈(98.4%)、甲醇(97.8%)、乙醇(94.3%)、丙酮(89.8%)、異丙醇(76.4%)、乙酸乙酯(62.3%)、二氯甲烷(57.1%),其中乙腈對4種黃酮類成分的提取率最高,但乙腈提取液中雜質較多,而甲醇對4種黃酮類成分的提取率為96.2%~102%,平均值為97.8%,甲醇提取液中雜質較少。試驗選擇提取溶劑為甲醇。
2.6 基質效應的評價
為考察黃芩提取物中目標物以外的基質對4種黃酮類成分提取效果的影響,在乙腈和空白黃芩提取物這兩種基質中分別加入5.0,100.0,250.0μg·kg-1的4種黃酮類成分混合標準溶液,采用標準加入法評估基質效應(即M=S2/S1×100%,其中:M為基質效應,S1為乙腈中目標物的質譜響應值,S2為黃芩提取物中目標物的質譜響應值。若90%<M<110%,表明不存在基質效應;若M<90%或M>110%時,表明存在基質減弱效應或基質增強效應)[15]。基質效應見表2。
由表2可知:M為92.7%~98.7%,即不存在基質效應。
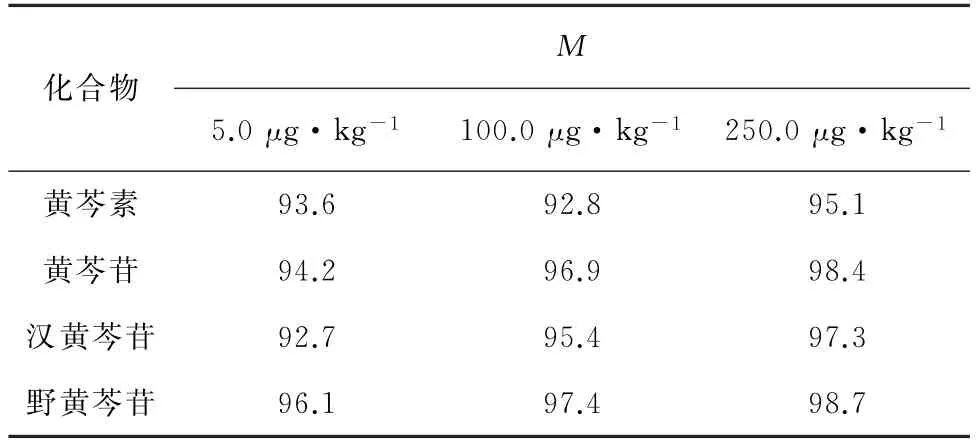
表2 基質效應Tab.2 Matrix effect %
2.7 標準曲線和檢出限
分別移取1 000 mg·L-1的4種黃酮類成分標準儲備溶液適量,用空白黃芩提取液為基質配制5.0,10.0,25.0,50.0,100.0μg·L-1的4種黃酮類成分的基質混合標準溶液系列。按儀器工作條件對上述基質混合標準溶液系列進行測定,以4種黃酮類成分的質量濃度為橫坐標,對應的質譜響應值為縱坐標,繪制標準曲線。結果表明:4種黃酮類成分的質量濃度在5.0~500.0μg·L-1內與其對應的質譜響應值呈線性關系,線性回歸方程和相關系數見表3。
根據3倍信噪比計算方法的檢出限(3S/N),根據10倍信噪比計算方法的測定下限(10S/N),結果見表3。
由表3可知:檢出限為0.3~1.5μg·kg-1,測定下限為1.0~5.0μg·kg-1。
2.8 精密度和回收試驗
按試驗方法對空白黃芩提取物樣品進行加標回收試驗,計算回收率和測定值的相對標準偏差(RSD),結果見表4。
由表4可知:回收率為91.1%~99.1%,RSD 為2.0%~4.1%。

表3 線性回歸方程、相關系數、檢出限和測定下限Tab.3 Linear regression equations,correlation coefficients,detection limits and lower limits of determination
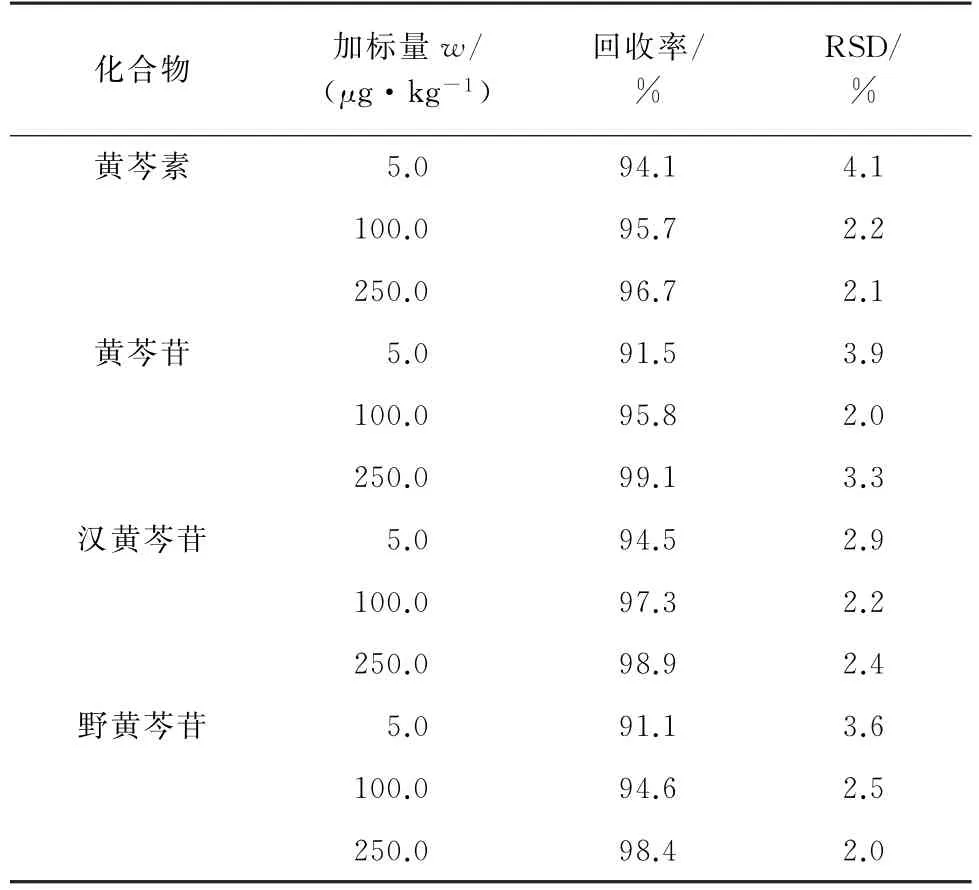
表4 精密度和回收試驗結果(n=6)Tab.4 Results of tests for precision and recovery(n=6)
2.9 樣品分析
按試驗方法對7份不同產地的黃芩提取物樣品進行分析,結果見表5。
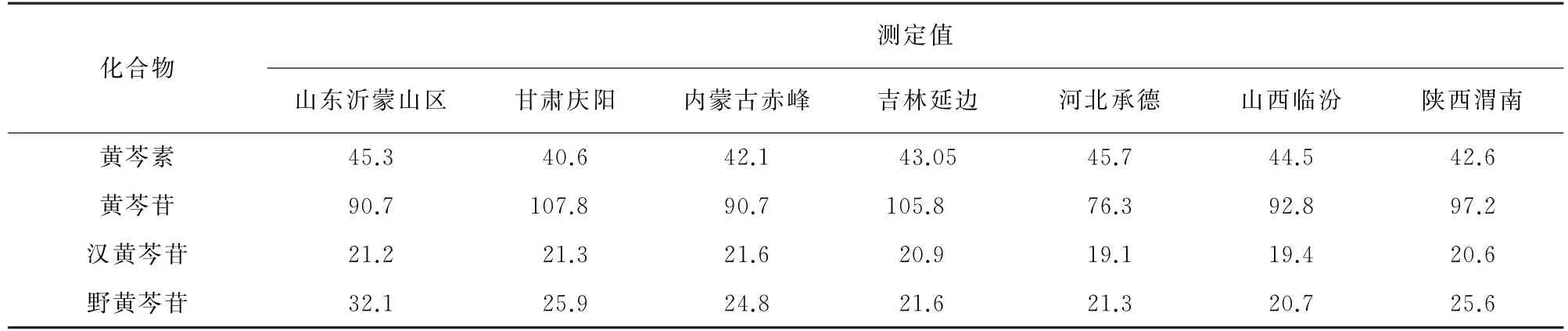
表5 樣品分析結果Tab.5 Analytical results of the samples mg·kg-1
由表5可知:不同產地的黃芩中均含有黃芩素、黃芩苷、漢黃芩苷、野黃芩苷,但其含量存在差異,這與文獻[1]、文獻[8]報道較為一致;其中黃芩苷含量差異較大,其質量分數為76.3~107.8 mg·kg-1,這種顯著性差異可能是由于黃芩品種、生長環境(氣溫、海拔、降雨量、日照等)、生長年限、貯藏時間及種植技術等差異引起的不同產地黃芩提取物之間相同的化學成分含量不同[16]。因此,利用黃芩提取物中黃芩苷、黃芩素、漢黃芩苷、野黃芩苷含量不同的特點,本方法可更加全面有效地為黃芩提取物質量控制提供科學數據。
本工作采用UHPLC-MS/MS測定黃芩提取物中黃芩素、黃芩苷、漢黃芩苷、野黃芩苷的含量。本方法具有靈敏度高、準確度好等特點。
