基于風險管理的藥品GMP 檢查發(fā)起機制研究
2020-07-11 05:44:04曹嘉成張書卉焦靈利
藥學與臨床研究 2020年3期
曹嘉成,張書卉,焦靈利
江蘇省食品藥品監(jiān)督管理局認證審評中心,南京210002
新修訂的《中華人民共和國藥品管理法》于2019 年12 月1 日起實施,規(guī)定取消對藥品生產企業(yè)的《藥品生產質量管理規(guī)范》(GMP)認證,改為對藥品生產企業(yè)是否符合生產質量管理規(guī)范開展檢查。《藥品管理法》的這一變化,意味著GMP 檢查由依企業(yè)申請變?yōu)楸O(jiān)管部門依風險發(fā)起,如何科學地判斷企業(yè)生產過程的風險,合理確定檢查范圍和檢查頻次,成為藥品監(jiān)管部門亟需思考解決的問題。藥品生產監(jiān)管風險較高,專業(yè)性較強,隨著藥品生產技術的日益精進,無菌保障要求不斷提高,單抗、CAR-T 等新產品不斷涌現,過去劃片區(qū)的網格化監(jiān)管,追求覆蓋率的痕跡化監(jiān)管已經不能滿足新時代的監(jiān)管形勢,需要省級監(jiān)管機構統(tǒng)籌謀劃、統(tǒng)一尺度、加強風險研判,制定科學的GMP 檢查發(fā)起機制,有針對性的開展檢查,控制風險。
1 我國藥品現行的GMP 檢查發(fā)起機制
1.1 藥品GMP 認證檢查
《藥品生產質量管理規(guī)范(2010 年修訂)》于2011 年3 月1 日起正式開始施行,要求所有血液制品、疫苗、注射劑等無菌藥品的生產應在2013 年12月31 日前達到GMP 要求,其他產品于2015 年之前全部達到GMP 要求。我國現行的GMP 按生產線進行認證,如一些企業(yè)有多條生產線,但對于企業(yè)本身而言,質量管理體系是一致的,同一劑型的不同車間設備及管理也幾乎相近,這類企業(yè)往往都是大型企業(yè),管理較完善,對于這種企業(yè)的反復認證是對行政資源的一種浪費。一些規(guī)模較小的企業(yè),往往只有1~2 個主要品種,由于經濟效益不高,這類企業(yè)往往人員素質相對較低,管理較薄弱,但這類企業(yè)只有到期換證才接受檢查,中間空檔期存在檢查缺失的風險。
1.2 藥品生產監(jiān)督檢查
在新版GMP 首輪基本完成后,監(jiān)管部門意識到GMP 認證檢查不能保持生產監(jiān)管的高壓態(tài)勢,國家局和省局逐步加強了監(jiān)督檢查的力度,對部分企業(yè)組織飛行檢查或跟蹤檢查,2016 年國家局對204家高風險企業(yè)進行了飛行檢查[1],但相對于全國近6000家藥品生產企業(yè)而言,只是冰山一角。各省級監(jiān)管部門每年也會對轄區(qū)內的企業(yè)開展有針對性的檢查,如江蘇局依2019 年對57家重點企業(yè)進行了檢查。
1.3 基于風險的檢查發(fā)起機制
僅僅通過產品本身的風險開展檢查,會造成一些綜合性生產企業(yè)被多次、重復檢查,對于合規(guī)情況較好的企業(yè),頻繁的檢查不僅是對行政資源的浪費,也給企業(yè)造成了一定的負擔。監(jiān)管部門應根據企業(yè)的劑型風險,結合企業(yè)歷史合規(guī)情況及近年來接受檢查的情況,制定一套基于風險的檢查發(fā)起機制,合理安排檢查資源,對部分高風險的企業(yè)加大檢查力度,同時應鼓勵企業(yè)自律,對一直以來合規(guī)情況較好的企業(yè),適當減少檢查頻次。
2 國外藥品生產檢查發(fā)起機制研究
2.1 美國藥品生產檢查發(fā)起機制
韓亮等[2]在《美國FDA 藥品生產質量監(jiān)管體系》中介紹,FDA 的藥品生產檢查主要分為3 種:一是新藥和仿制藥申請的批準前檢查;二是常規(guī)GMP檢查;三是有質量投訴或者突發(fā)事件等的有因檢查。藥品上市后的檢查主要包括常規(guī)GMP 檢查和有因檢查,FDA 并沒有明確規(guī)定現場檢查的有效期,而會根據藥品生產企業(yè)風險評估模型,確定檢查發(fā)起的頻次。FDA 建立的藥品生產風險評估模型主要從產品本身、質量體系和設施設備情況、生產中操作不當三個方面分析風險要素[3],給其進行賦值,根據得分結果評價企業(yè)風險高低,對風險大的企業(yè)加強監(jiān)督,而對風險小的企業(yè)則降低檢查強度。
2.2 歐盟國家藥品生產檢查發(fā)起機制
歐盟的藥品監(jiān)管主要由European Medicines Agency(EMA)與各成員國自身的藥品監(jiān)管機構組成,EMA 主要負責藥品的集中注冊審批工作,各成員國的藥品檢查機構負責各自區(qū)域的GMP 檢查工作[4]。在德國,食品、藥品的日常監(jiān)管基本都屬于各地方政府的職權。各地對生產和銷售企業(yè)的檢查是不定期的,基本采取飛行檢查的方式。檢查的頻率取決于兩大因素:一是企業(yè)產品本身的風險程度,例如注射劑等無菌制劑就要比片劑等普通固體藥品檢查頻率高;二是依據歷次的合規(guī)情況,如一個企業(yè)前10 次檢查均合規(guī)情況良好,以后對其檢查的頻率就會降低,反之就升高[5]。
2.3 澳大利亞藥品生產檢查發(fā)起機制
澳大利亞治療產品管理局(Therapeutic Goods Administration,TGA),對澳大利亞藥品和醫(yī)療器械的質量、安全、有效性和供應負責,聯(lián)邦和州政府都設有藥品監(jiān)管機構,負責藥品生產的日常檢查。TGA制定檢查頻次主要依據企業(yè)產品風險和歷年來合規(guī)檢查情況,如無菌產品初始合規(guī)檢查每24 個月一次,如果連續(xù)3 次合規(guī)情況良好,則36 個月檢查一次[6]。
3 藥品生產風險要素分析
基于國外風險評估的原則,綜合考慮我國現階段檢查發(fā)起實際,藥品生產風險評估模型主要分為三部分:一是企業(yè)自身風險;二是企業(yè)合規(guī)歷史;三是接受檢查頻次。
3.1 企業(yè)自身風險
藥品生產企業(yè)的風險主要來源于四個方面:一是企業(yè)經營的情況;二是企業(yè)生產品種的風險;三是企業(yè)人員素質;四是企業(yè)設施設備的情況。對上述風險涉及的具體風險指標進行分析,見表1。

表1 企業(yè)自身風險
3.2 企業(yè)合規(guī)歷史 見表2。
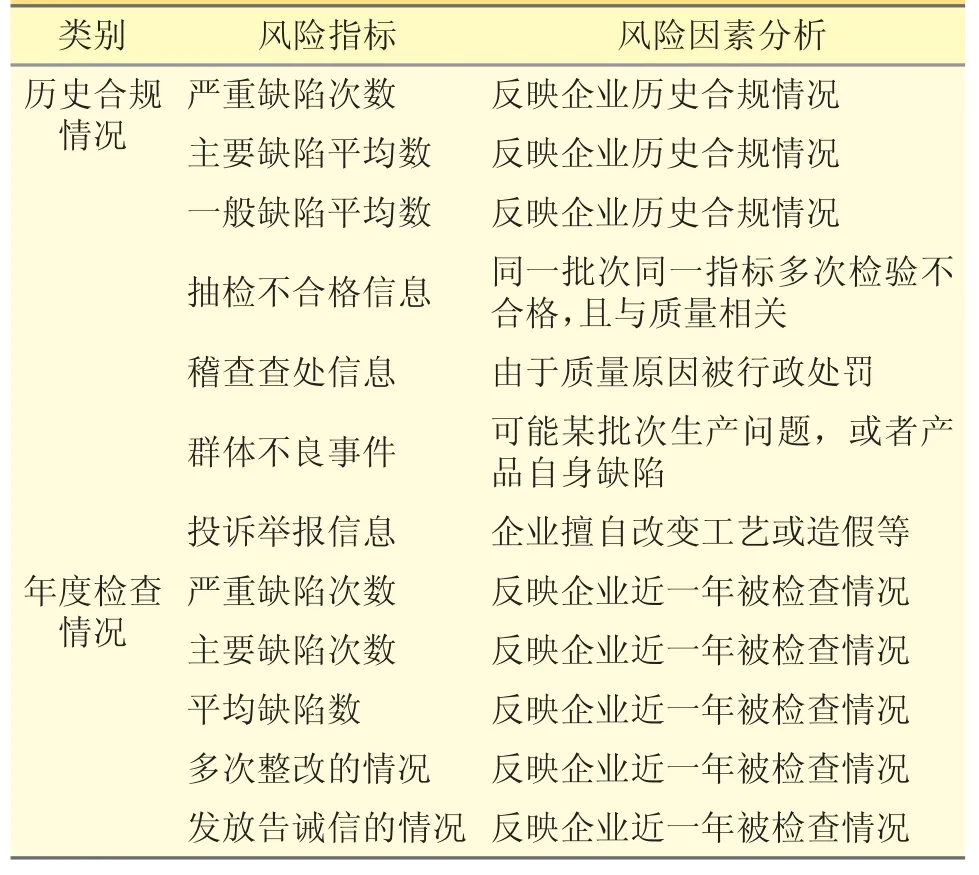
表2 企業(yè)合規(guī)歷史
3.3 接受檢查頻次 見表3。
4 藥品生產風險評估模型建立與試算
根據上述風險指標的分析,邀請業(yè)內資深檢查員及企業(yè)質量管理人員對各風險指標進行分析,對風險指標的高低進行排序,根據排序情況對各指標進行合理賦值。賦值內容:企業(yè)自身風險;企業(yè)合規(guī)歷史;接受檢查頻次及評分方法。
4.1 企業(yè)自身風險賦值 見表4。
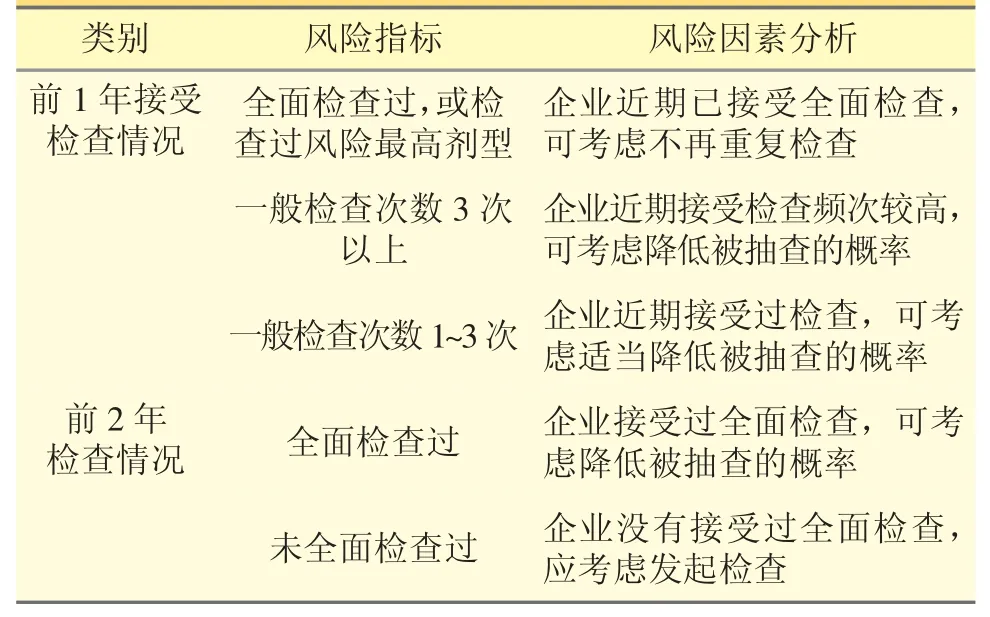
表3 接受檢查頻次
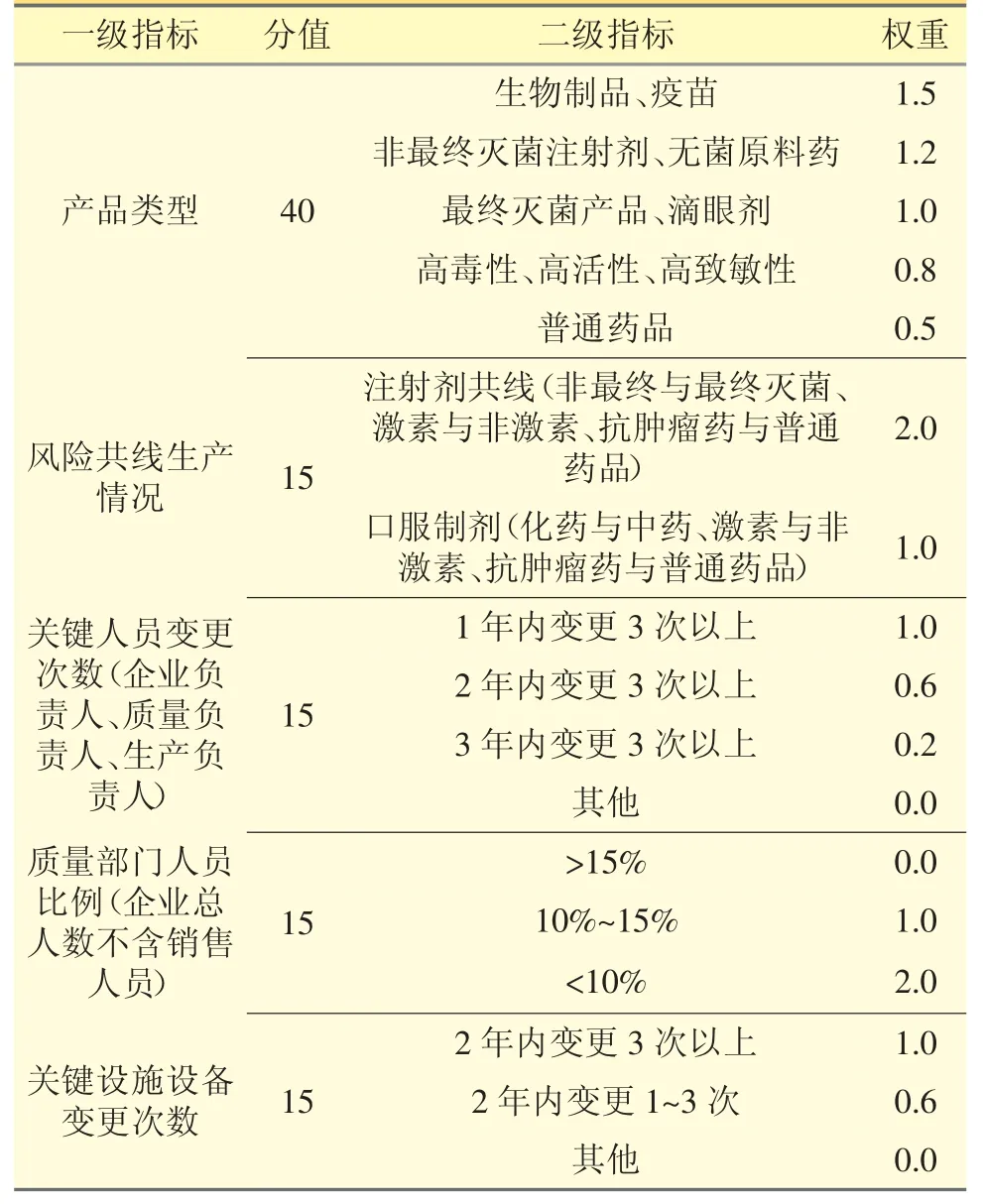
表4 企業(yè)自身風險賦值
4.2 企業(yè)合規(guī)歷史賦值 見表5。
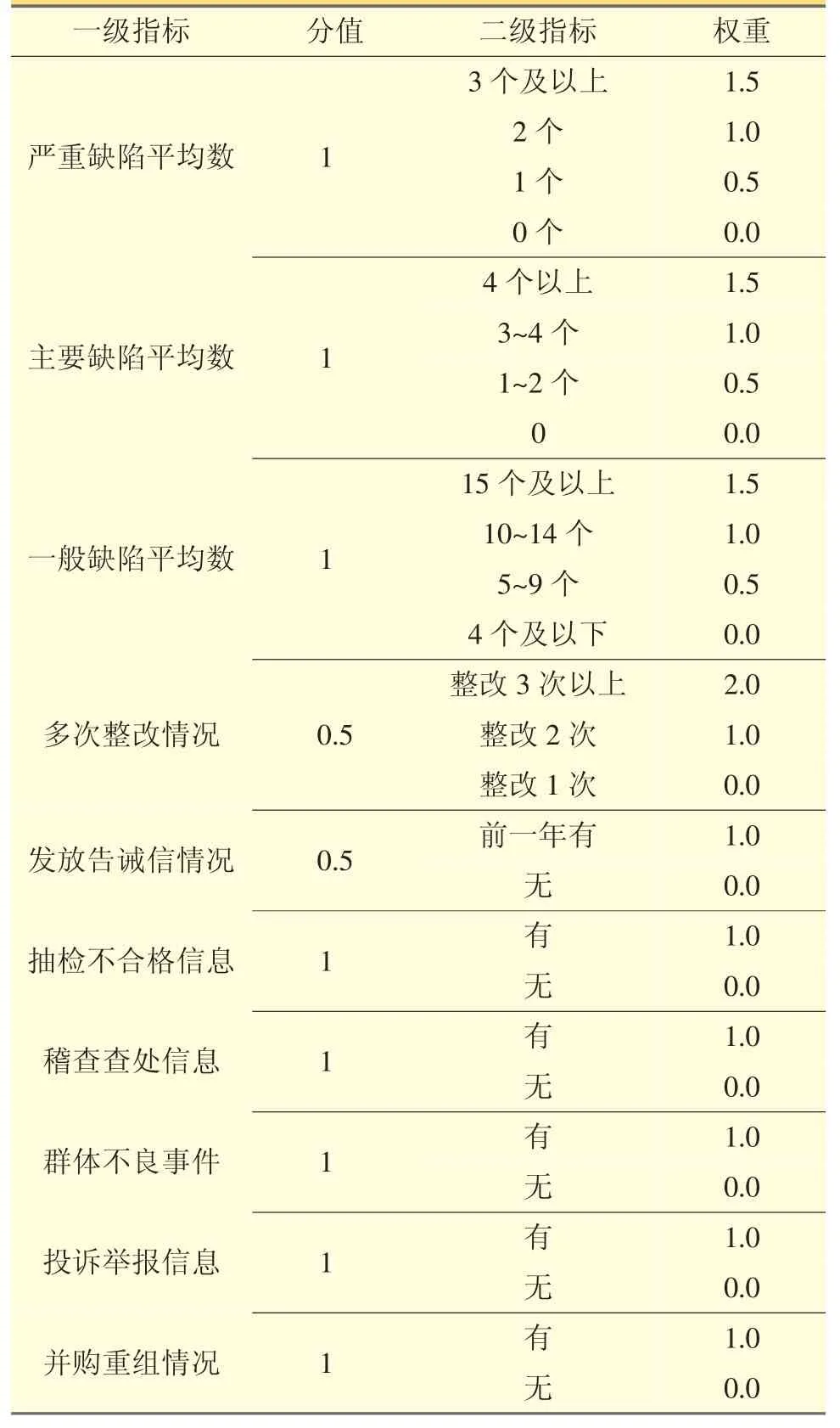
表5 企業(yè)合規(guī)歷史賦值
4.3 接受檢查頻次賦值 見表6。

表6 接受檢查頻次賦值
4.4 評分方法
采用動態(tài)加權綜合評價方法。將“企業(yè)自身風險”、“企業(yè)合規(guī)歷史”、“接受檢查頻次”三個評價系統(tǒng)分別記為S1、S2、S3,每個系統(tǒng)的具體指標分為一級指標分值(X1、X2、…、Xm)和二級指標權重(P1、P2、…、Pm),打分=企業(yè)自身風險{∑(一級指標分值×二級指標權重)}×企業(yè)合規(guī)歷史{∑(一級指標分值×二級指標權重)}×接受檢查頻次{∑(一級指標分值×二級指標權重)}。
4.5 風險評估模型試算
選取本省4家藥品生產企業(yè)進行模型試算,見表7。
5 藥品生產風險評估模型動態(tài)調整
藥品生產企業(yè)的風險并不是固定不變的,企業(yè)的經營、品種、人員、設施設備等情況都在隨時變化。風險指標的賦值主要根據監(jiān)管需求和監(jiān)管經驗而定,賦值需要考慮多個因素:一是風險指標并不是孤立的,內在有著充分的聯(lián)系,如企業(yè)經營情況不善,可能會導致人員流動性較大;二是風險具有兩面性,如設施設備的更新可能會帶來工藝改變或者驗證不充分的風險,但是從長遠考慮,是企業(yè)良性發(fā)展的必由之路;三是監(jiān)管風險與企業(yè)風險的區(qū)分,如無菌制劑監(jiān)管風險較高,但是如果企業(yè)無菌保障能力強,質量管理水平高,風險控制能力強,實際的監(jiān)管風險并不高。生產風險指標的賦值不僅需要綜合各方面因素,還要根據實際情況動態(tài)調整,既要根據監(jiān)管經驗,又要結合監(jiān)管的新形勢、新要求,同時還要考慮行業(yè)發(fā)展的動態(tài)。
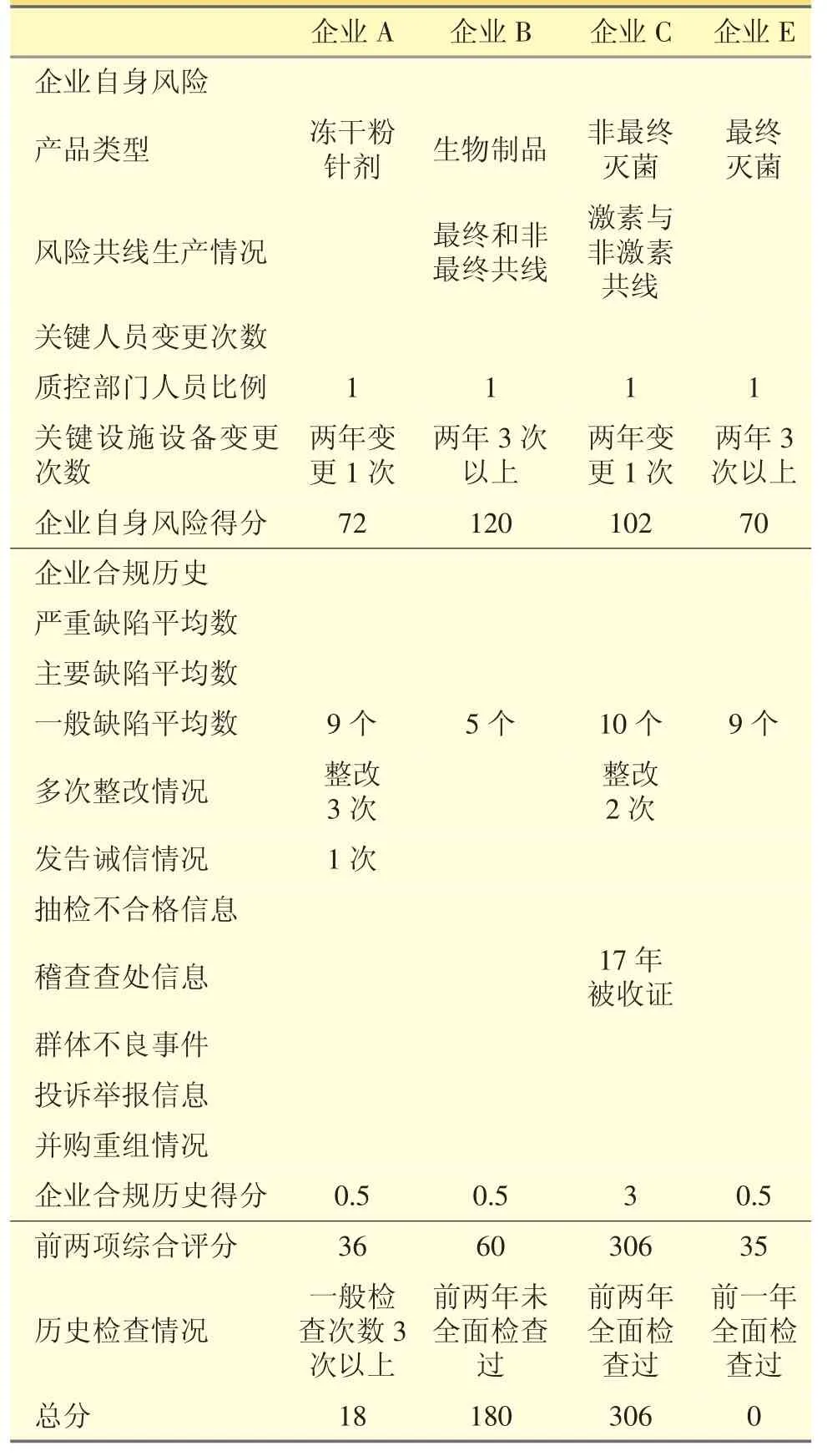
表7 江蘇4家藥品生產企業(yè)風險評估模型試算
監(jiān)管風險指標賦值及權重應由資深檢查員及監(jiān)管人員根據檢查經驗和檢查工作量擬定,通過檢查的大數據反映的檢查績效來驗證風險評估模型的合理性。同時,建立評分的動態(tài)調整機制,模型的評分根據檢查績效、監(jiān)管需求、行業(yè)動態(tài)進行合理調整,保證風險評估模型的時效性、檢查的指向性、監(jiān)管的科學性。
6 結語
新修訂的《藥品管理法》取消了GMP 認證方式,由依照申請檢查改為依照風險檢查,本文利用風險評估工具及統(tǒng)計學手段,建立了藥品生產風險評估模型,提高了GMP 檢查的指向性,為GMP 檢查新方式提供了科學依據,為合理利用行政資源提供有力抓手,為藥品監(jiān)管科學可持續(xù)發(fā)展、加快推動藥品監(jiān)管治理體系和治理能力現代化提供思路。
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
當代水產(2022年5期)2022-06-05 07:55:06
當代水產(2022年3期)2022-04-26 14:27:04
當代水產(2022年2期)2022-04-26 14:25:10
小學科學(學生版)(2020年10期)2020-10-28 07:52:12
云南畫報(2020年9期)2020-10-27 02:03:26
中國化肥信息(2020年7期)2020-03-19 01:54:02
中國軍轉民(2017年6期)2018-01-31 02:22:28
中國衛(wèi)生(2016年5期)2016-11-12 13:25:28
中國衛(wèi)生(2015年5期)2015-11-08 12:09:48
