RP-HPLC 法測定藥用膠塞中16 種多環(huán)芳烴含量并作模擬提取研究
2020-07-11 05:43:58張曉蕓劉露遙丁逸梅
藥學(xué)與臨床研究 2020年3期
張曉蕓,劉露遙,丁逸梅
江蘇省藥物研究所有限公司,南京210009
多環(huán)芳烴(polycyclic aromatic hydro-carbons,PAHs)是一類由兩個或兩個以上苯環(huán)以稠環(huán)形式相連的有機化合物,主要包含萘、苊烯、芴、菲、蒽、熒蒽、芘、苯并蒽、艸屈、苯并[b]熒蒽、苯并[k]熒蒽、苯并[a]芘、茚[1,2,3-cd]并芘、二苯并[a,h]蒽和苯并苝等。含有四到六個環(huán)的稠環(huán)多環(huán)芳烴具有致癌性,1976年國際癌癥研究中心(IARC)公布了94 種動物致癌物質(zhì)[1],其中多環(huán)芳烴化合物含有15 種,以苯并[α]芘為首個環(huán)境化學(xué)強致癌物質(zhì);多環(huán)芳烴具有很強的脂溶性,較難降解,并且容易在生物體內(nèi)蓄積。到目前為止,歐盟2005/69/EC 指令(即76/769/EEC 之第27 次修訂版)規(guī)定橡膠油中苯并[α]芘不得超過1mg·kg-1。2005 年9 月德國發(fā)布的《德國食品和商品法》中規(guī)定,PAHs 總量的最大允許限量是10 mg·kg-1,苯并[α]芘最大允許限量是1 mg·kg-1;美國和中國等各國均通過法律或法令對多環(huán)芳烴的含量有明確的限量要求[1,2]。橡膠中多環(huán)芳烴存在于膠塞生產(chǎn)原料橡膠油中,橡膠油作為一種軟化劑可以使橡膠具有良好的彈性和韌性。注射劑等藥物常見的包裝材料為橡膠膠塞,如果膠塞中殘留的PAHs 遷移至藥液中會對患者造成危害;通常選擇藥用膠塞的材質(zhì)分為:注射用冷凍干燥無菌粉末覆聚四氟乙烯/乙烯共聚物膜氯化丁基膠塞和注射用鹵化丁基橡膠塞(氯化或者溴化)。目前,多環(huán)芳烴的檢測技術(shù)主要有化學(xué)滴定法、高效液相色譜法、氣相色譜-質(zhì)譜聯(lián)用法、氣相色譜法、電化學(xué)法、分光光度法及熱透鏡光度法、拉曼光譜分析法等[3-13]。其中在環(huán)境保護領(lǐng)域較為常見的主要有高效液相色譜法、氣相色譜-質(zhì)譜聯(lián)用法。本文采用RP-HPLC 法測定藥用膠塞中16 種多環(huán)芳烴的含量,并作其遷移試驗。
1 實驗部分
1.1 儀器與材料
儀器:高效液相色譜儀LC-2010CHT(編號:C21255010791,日本島津公司);電子分析天平(BSA224S,賽多利斯科學(xué)儀器(北京)有限公司);手提式壓力蒸汽滅菌器(上海劃線醫(yī)用核子儀器有限公司,編號:428)。
16 種多環(huán)芳烴混合對照品(PAHs-Solution16-22),來源:O2Si,批號:320418,質(zhì)量濃度(萘1002 mg·L-1,苊2002 mg·L-1,茐200.1 mg·L-1,二氫苊999.9 mg·L-1,菲100.2 mg·L-1,蒽100.2 mg·L-1,熒蒽200.1 mg·L-1,芘99.84 mg·L-1,艸屈100.6 mg·L-1,苯并[a]蒽100.4 mg·L-1,苯并[b]熒蒽200.1 mg·L-1,苯并[k]熒蒽100.4 mg·L-1,二苯并[a]芘100.7 mg·L-1,二苯并[ah]蒽199.9 mg·L-1,苯并[ghi]苝200 mg·L-1,茚并[1,2,3-cd]芘99.96 mg·L-1)。16 種多環(huán)芳烴混合對照品(PAHs-Mix-EPA610),來源:O2Si 標準品公司,批號:300821,質(zhì)量濃度均為200 μg·mL-1;萘對照品,來源:阿拉丁試劑公司,批號:C1819049,純度98%。
藥用膠塞:①注射用冷凍干燥無菌粉末用覆聚四氟乙烯/乙烯共聚物膜氯化丁基膠塞,來源:江蘇華蘭藥用新材料股份有限公司,批號:M170406122-78;②藥用鍍聚四氟乙烯膜氯化丁基橡膠/聚異戊二烯橡膠塞,來源:新加坡西式西藥服務(wù)公司,批號:3138 010040;③注射用鹵化丁基橡膠塞(溴化),來源:江蘇博生醫(yī)用新材料股份有限公司,批號:16 102362;④注射用冷凍干燥無菌粉末氯化丁基橡膠塞,來源:江蘇華蘭藥用新材料股份有限公司,批號:170508159-60。
藥品與試劑:注射用蘭索拉唑,來源:江蘇某藥業(yè)股份有限公司,批號:180301,180302,180303;注射用帕瑞希布鈉,來源:杭州某藥業(yè)有限公司,批號:19010814,19010914,19 011014;注射用萬古霉素,來源:浙江某藥業(yè)股份有限公司,批號:20170901,20170902,20 170903;乙腈,來源:Honeywell,批號:S3ZA1H);乙醇、正己烷為分析純;實驗用水為重蒸水。
1.2 方法
1.2.1 色譜條件 色譜柱:Kromasil 100-5-C18(4.6 mm×250 mm,5 μm);流速:1.2 mL·min-1;柱溫:35 ℃;檢測波長:220(萘、苊、二氫苊)、254(茐、菲、蒽、熒蒽、芘、艸屈、苯并[a]蒽、苯并[b]熒蒽、苯并[k]熒蒽、二苯并[a]芘、苯并[ghi]苝)、295(二苯并[ah]蒽、茚并[1,2,3-cd]芘)nm;進樣量:10 μL。流動相:色譜純乙腈、屈臣氏蒸餾水(采用梯度洗脫:0~27 min,乙腈65%;27~41 min,乙腈65%~100%;41~43 min,乙腈100%~65%;43~55 min,乙腈65%)。16 種PAHs 對照品的典型圖譜見圖1。
1.2.2 氣相色譜條件 Thermo Scientific Trace GC Ultra-ISQ 氣質(zhì)聯(lián)用儀,軟件:Xcalibur;離子源:EI;色譜柱:Agilent DB-5 MS UI(30m×250μm×0.25μm);進樣量:1.5 μL;不分流;恒定流量:1.2 mL·min-1;進樣口溫度:280 ℃;柱溫:60 ℃,保持1 min,以10 ℃·min-1升至110 ℃,保持2 min,以5 ℃·min-1升至300 ℃,保持10 min;傳輸線溫度:250 ℃;離子源溫度:250 ℃;溶劑延遲:6.0 min;定量離子搜索模式(SIM)。定性與定量離子選擇見表1。
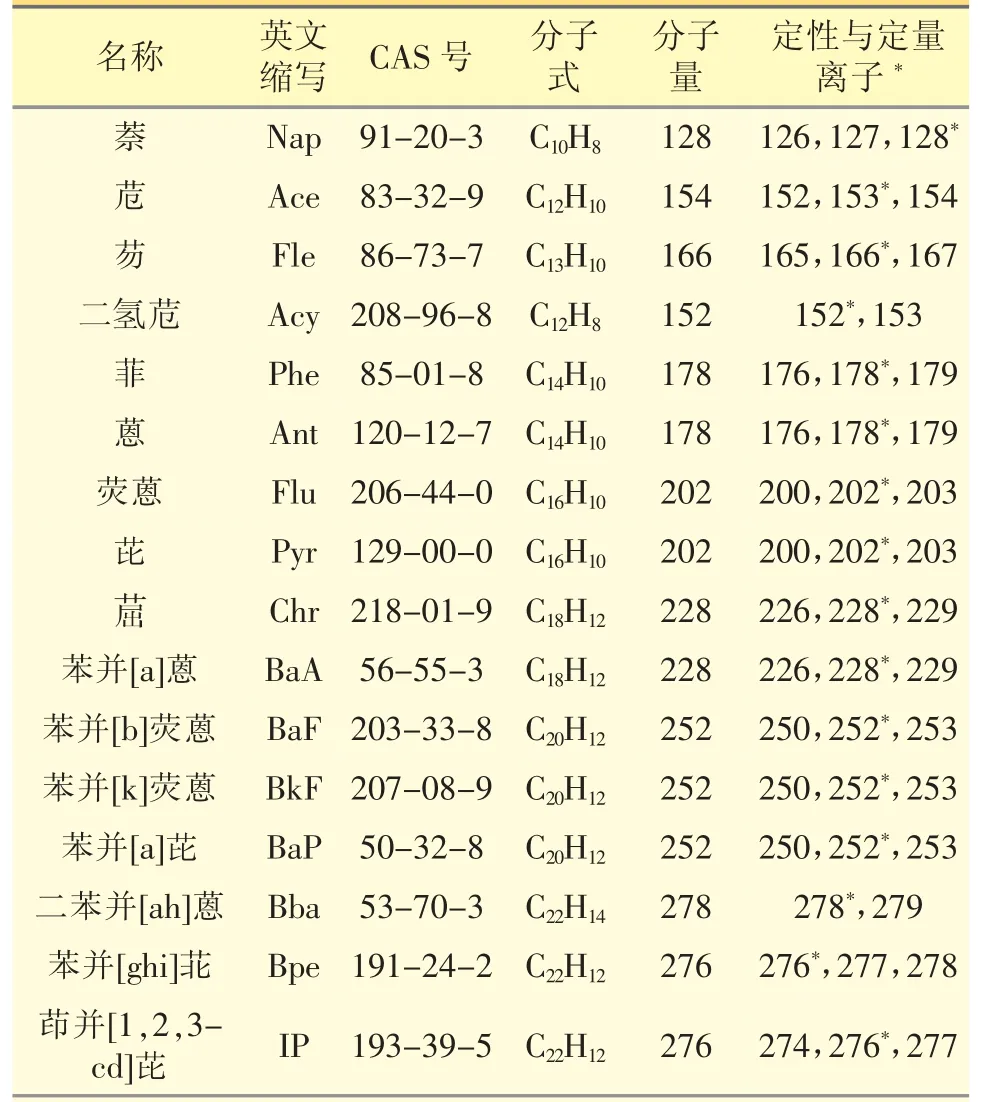
表1 定性與定量離子選擇
1.2.3 提取試驗 膠塞經(jīng)121 ℃高壓滅菌15 min后,取出剪碎,稱取約10 g,精密稱定,置100 mL 錐形瓶中,加20 mL 無水乙醇,50 ℃超聲4 h;取出冷卻,取上清液即為提取液。
1.2.4 遷移試驗
1.2.4.1 對照品溶液制備 精密量取PAHs 混合對照品適量,至100 mL 量瓶中,用甲醇稀釋成PAHs含量為0.1 μg·mL-1的混合對照品溶液,待用。
1.2.4.2 樣品溶液制備 ①取注射用蘭索拉唑(批號:180301,180302,180303)凍干粉末3 瓶,模擬臨床使用,每瓶加入5 mL 0.9% NaCl,混合均勻;精密量取10 mL 該樣品溶液,精密加入5 mL 正己烷,渦旋5 min,靜置分層,精密量取有機層1.0 mL,用高純氮氣吹干后,加入1.0 mL 無水乙醇復(fù)溶。②取注射用帕瑞希布鈉(批號:19010814,19010914,19011014)凍干粉末,模擬臨床使用每瓶加入5mL 0.9% NaCl,取2 瓶混合均勻后備用;精密量取5.0mL 該樣品溶液,加入5 mL 正己烷,渦旋5 min,靜置分層,精密量取有機層1.0mL,用高純氮氣吹干后,精密加入1.0mL無水乙醇復(fù)溶。③取注射用萬古霉素(批號:20170901,20170902,20170903)凍干粉末,模擬臨床使用每瓶加入5 mL 0.9% NaCl,取2 瓶混合均勻后備用;精密量取5.0 mL 該樣品溶液,加入5.0 mL 正己烷,渦旋5 min,靜置分層,精密量取有機層1.0 mL,用高純氮氣吹干后,精密加入1.0 mL 無水乙醇復(fù)溶。
1.3 結(jié)果
1.3.1 提取實驗
1.3.1.1 線性范圍 精密量取PAHs 混合對照品溶液適量,用乙腈逐級稀釋,制備線性溶液,濃度分別為0.1000、0.500、1.000、5.000、10.00 μg·mL-1;精密量取上述對照品溶液10 μL,注入液相色譜儀測定,以對照品濃度為橫坐標,峰面積為縱坐標,繪制標準曲線。結(jié)果表明,各PAHs 在0.1000~10.00 μg·mL-1范圍內(nèi)線性關(guān)系良好,相關(guān)系數(shù)(r)≥0.999。以信噪比S/N=3 為最低檢出限,檢出限0.751 5 ng·mL-1~24.96 ng·mL-1;以信噪比S/N=10 為最低定量限2.505 ng·mL-1~83.20 ng·mL-1。見表2。
1.3.1.2 進樣精密度試驗 精密量取對照品溶液10 μL,注入液相色譜儀測定,連續(xù)進樣5 次,測得峰面積的RSD 均≤2.0%,表明進樣精密度良好。
1.3.1.3 回收率實驗 取“1.2.3”項的膠塞10 g,共9份,使用含有濃度為0.1000、0.5000、1.000μg·mL-1的PAHs 對照品溶液20mL,按照“1.2.3”項下方法制備膠塞樣品溶液,精密量取該溶液10μL,注入液相色譜儀測定,以外標法計算得到回收率為88.74%~99.27%。
1.3.1.4 重復(fù)性試驗 取“1.2.3”項的膠塞10 g,共6 份,精密加入濃度為0.5000 μg·mL-1LPAHs 混合對照品溶液20 mL,按照“1.2.3”項下方法制備膠塞樣品溶液,精密量取該溶液10.0 μL,注入液相色譜儀測定,測得的RSD≤3.0%。
1.3.1.5 膠塞提取 對市售的藥用膠塞進行PAHs的檢測,每種膠塞按照“1.2.3”項下方法制備膠塞樣品溶液。萘、苊和二氫苊的檢測波長為220 nm,依法測定,由圖2A 可知,膠塞中可能存在萘,提取液圖譜中苊的保留時間與對照品圖譜中的苊的保留時間不一致,待GCMS 進行分子量鑒定;茐、菲、蒽、熒蒽、芘、艸屈、苯并[a]蒽、苯并[b]熒蒽、苯并[k]熒蒽、二苯并[a]芘和苯并[ghi]苝的檢測波長為254 nm,由圖2B 可知,該類化合物均未檢出;二苯并[ah]蒽和茚并[1,2,3-cd]芘的檢測波長為295 nm,由圖2C 可知,這兩種化合物均未檢出。其提取液HPLC 圖譜見表3。
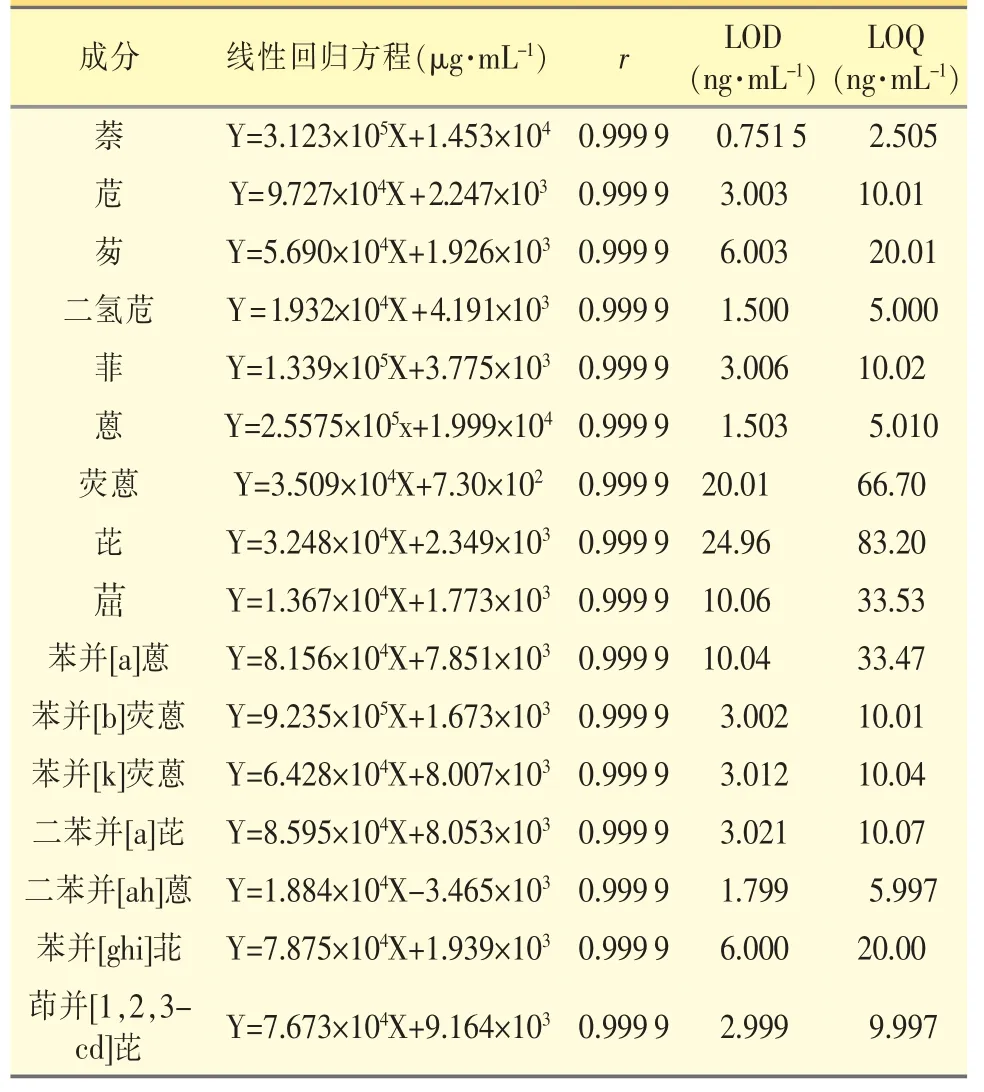
表2 16 種PAHs 的回歸方程
1.3.2 遷移實驗
1.3.2.1 萃取溶劑篩選 本實驗篩選了溶劑:乙醚、乙酸乙酯、二氯甲烷、三氯甲烷、正己烷,乙醚沸點太低,難以精密量取;乙酸乙酯、二氯甲烷、三氯甲烷對主藥部分溶解,干擾多環(huán)芳烴的檢測;多環(huán)芳烴在正己烷中溶解性較好,主藥及雜質(zhì)在正己烷中溶解性較差,使用正己烷提取干擾較少,回收率高。
1.3.2.2 回收率試驗 分別取注射用蘭索拉唑樣品(批號180301)、注射用帕瑞昔布鈉(批號19010814)和注射用萬古霉素樣品(批號20170901),精密加入含PAHs 濃度為0.1000、0.5000、1.000 μg·mL-1的乙醇溶液,按照“1.2.4.2”方法制備樣品溶液;精密量取該溶液10 μL,注入液相色譜儀,記錄各色譜峰峰面積,采用外標法計算回收率及RSD 值,注射用蘭索拉唑平均加樣回收率為94.28%~98.81%,RSD≤5.0%;注射用帕瑞昔布鈉平均加樣回收率為93.61%~99.43%,RSD≤5.0%;注射用萬古霉素平均加樣回收率為93.34%~98.89%,RSD≤5.0%。見表4。

表3 膠塞多環(huán)芳烴提取結(jié)果(ng·g-1)
1.3.2.3 注射液中PAHs 的測定 ①選取3 批注射用蘭索拉唑樣品(批號:180301,180302,180303),進行PHAs 遷移試驗,按照“1.2.4.2”項下方法制備樣品溶液;精密量取該溶液10.0 μL,注入液相色譜儀測定,3 批樣品中均未檢出PHAs。②取3 批注射用帕瑞希布鈉(批號:19010814,19010914,19011014),進行PHAs 遷移試驗,按照“1.2.4.2”項下方法制備樣品溶液;精密量取該溶液10.0 μL,注入液相色譜儀測定,3 批樣品中均未檢出PHAs。③取3 批注射用萬古霉素(批號:20170901,20170902,20170903)進行PHAs 遷移試驗,按照“1.2.4.2”項下方法制備樣品溶液;精密量取該溶液10.0 μL,注入液相色譜儀測定,3 批樣品中均未檢出PHAs。
2 結(jié)論
本實驗使用高效液相色譜法測定藥用膠塞中16 種PAHs,各PAH 范圍在0.100 0~10.00 μg·mL-1濃度之間內(nèi),其峰面積與濃度線性關(guān)系良好,相關(guān)系數(shù)r≥0.999;檢測限0.751 5~24.96 ng·mL-1;定量限2.505~83.20 ng·mL-1;精密度的RSD≤2.0%;重復(fù)性的RSD≤3.0%;平均加樣回收率為88.74%~99.27%。
經(jīng)GCMS 定性分析,表明膠塞中提取物為萘,目前GCMS 也能夠?qū)AHs 良好分離,且靈敏度良好;但考慮GCMS 自身的缺陷性,適用于定性和半定量分析,且GCMS 相對成本較高。本文仍采用液-液萃取的方式,以HPLC-UV 法考察PAHs 的遷移實驗。
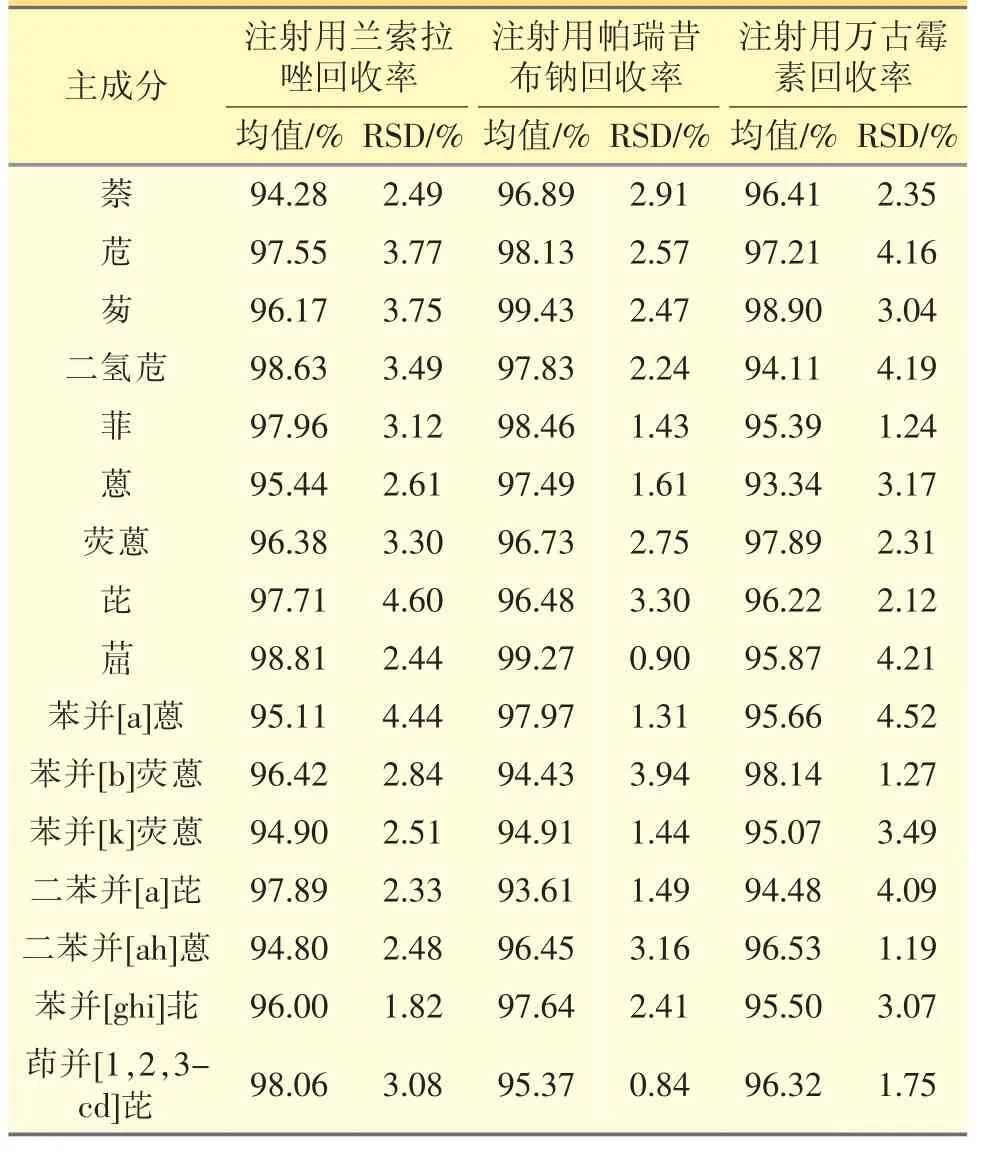
表4 注射用蘭索拉唑、帕瑞昔布鈉和萬古霉素中PAHs 的遷移平均回收率及RSD 值
本試驗選用注射用蘭索拉唑等3 個樣品為模型注射劑,建立了16 種PAHs 的遷移實驗,通過加標方式考察該方法的專屬性,注射用蘭索拉唑平均加樣回收率為94.28%~98.81%,RSD≤5.0%;注射用帕瑞昔布鈉平均加樣回收率為93.61%~99.43%,RSD≤5.0%;注射用萬古霉素平均加樣回收率為93.34%~98.90%,RSD≤5.0%。本試驗采用的HPLC法測定藥用膠塞中的PAHs,方法良好,能夠快捷、簡便、準確地檢測出橡膠膠塞中16 種PAHs 的含量,適用于橡膠膠塞中PAHs 的測定。通過溶劑篩選得到正己烷為注射劑藥液的合適的萃取溶劑,能夠準確考察注射液中16 種PAHs 的遷移。
