HPLC-PAD法測定西洋參類保健食品中10種皂苷的含量
2020-07-06 06:36:46吳曉云刁飛燕李秀慧劉春霖李啟艷
藥學研究 2020年6期
吳曉云,刁飛燕,李秀慧,劉春霖,李啟艷
(山東省食品藥品檢驗研究院,山東 濟南 250101)
西洋參為五加科人參屬植物,是名貴的中藥材,人參皂苷是其主要活性成分,主要有人參皂苷Rg1、Rb1、Rb2、Rc、Rd和Re等。以西洋參為原料的保健食品具有緩解體力疲勞,增強免疫力、抗氧化和抗腫瘤等作用[1]。目前,西洋參類保健食品的劑型有硬膠囊、軟膠囊、片劑和口服溶液等,主要以總皂苷作為標志性成分,總皂苷的測定主要采用香草醛-高氯酸或硫酸顯色后用紫外分光光度法測定[2],該方法存在專屬性差,操作復雜和干擾因素多等缺點。
為此,徐燦輝等[3]改進了西洋參類保健食品中人參皂苷測定方法,建立了西洋參類保健食品中7種參皂苷含量高效液相色譜(HPLC)測定的方法。此外,人參皂苷測定方法還有超高效液相色譜(UPLC)[4]、高效液相色譜-質譜聯用法(HPLC-MS)[5-6]等。在眾多資料中,主要研究西洋參根莖葉提取物中人參皂苷含量,但對西洋參類保健食品中10種人參皂苷含量測定的報道較少。
本試驗通過參考西洋參藥材中皂苷測定的有關文獻[7-9],建立高效液相色譜法同時測定多種劑型西洋參類保健食品中10種人參皂苷,為質量標準的提升提供依據。
1 試驗部分
1.1 儀器 液相色譜儀(Agilent 1260 高效液相色譜儀,美國安捷倫公司), 配二極管陣列檢測器(PAD);電子天平(Mettler Toledo MS,梅特勒-托利多);數控超聲波清洗器(KQ-500DE型,昆山市超聲儀器有限公司);恒溫水浴鍋(北京永光明)。
1.2 試藥與供試品 乙腈(色譜純,Honeywell);甲醇(色譜純,Honeywell);超純水;正丁醇(分析純,國藥集團);氨水(分析純,國藥集團)。
標準品:人參皂苷Rb1、Rb2、Rb3、Rg1、Rg3、Rd、Re由中國食品藥品檢定研究院提供,含量分別為95.9%、93.8%、97.0%、96.3%、100%、94.4%、97.4%,人參皂苷Rg2、Rc、Rf由上海甄準生物科技有限公司提供,含量分別為98.02%、99.11%、99.62%。
供試品均由市場購得,名稱與劑型見表1。

表1 12種供試品的名稱和劑型
2 方法與結果
2.1 色譜條件 色譜柱:Kromasil C18(4.6 mm×250 mm,5 μm);流動相:乙腈(A)-水(B),梯度洗脫(0~40 min,17%A→19%A;40~60 min,19%A→29%A;60~75 min,29%A;75~100 min,29%A→40%A;100~105 min,40%A→17%A);流速1.0 mL·min-1;檢測波長203 nm;柱溫35 ℃;進樣量:10 μL。
2.2 對照品儲備液及對照品混合工作液配制 分別精密稱定人參皂苷Rg1、Rg2、Rg3、Rb1、Rb2、Rb3、Rc、Rd、Re、Rf對照品適量,置于25 mL量瓶中,用甲醇溶解并定容,制成人參皂苷單體濃度分別為2.409、2.141、0.047 12、1.947、1.758、2.138、2.250、2.062、2.077、2.008 mg·mL-1的對照品儲備液。分別取10種人參皂苷對照品儲備液適量,加甲醇稀釋制成6個濃度的混合對照品工作液。
2.3 供試品溶液的制備
2.3.1 片劑、膠囊劑供試品溶液的制備 片劑、膠囊劑,取內容物研磨混勻后,片劑2 g,膠囊劑1 g,精密稱定,置于100 mL錐形瓶中,精密加水飽和正丁醇50 mL,密塞,放置過夜,超聲處理(功率250 W,頻率50 kHz)30 min,濾過,棄去初濾液,精密量取續濾液20 mL,用氨試液洗滌兩次,每次20 mL,正丁醇提取液蒸干后,殘渣加甲醇適量使溶解,作為供試品溶液。
2.3.2 口服溶液供試品溶液的制備 口服溶液,精密量取8.0 mL供試品至分液漏斗中,用水飽和正丁醇振搖提取3次,每次10 mL,合并正丁醇提取液,用氨試液洗滌2次,每次10 mL,正丁醇提取液蒸干后,殘渣加甲醇適量使溶解,作為供試品溶液。
2.4 線性關系考察 分別取6個濃度的混合對照品工作液,進樣10 μL,記錄峰面積,以對照品濃度X(μg·mL-1)為橫坐標,對照品的峰面積Y為縱坐標,繪制標準曲線,求得回歸方程。得到10種人參皂苷在相應線性范圍內均具有良好的線性,相關系數都在0.99以上,結果見表2。
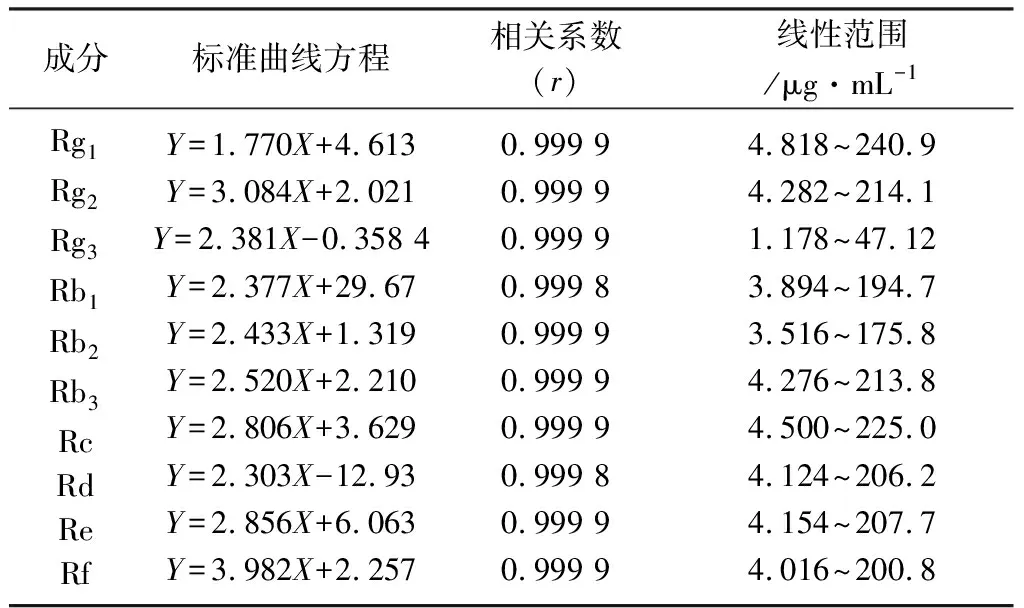
表2 標準曲線方程的結果
2.5 試樣重復性試驗 準確量取6份口服溶液供試品(S01)8.0 mL至分液漏斗中,以下按“2.3.2”項下方法操作,制備供試品溶液。
準確稱取6份膠囊劑供試品(S05)1 g,6份片劑供試品(S08)2 g,置于100 mL錐形瓶中,以下按“2.3.1”項下方法操作,制備供試品溶液。
分別取3種劑型供試品溶液10 μL注入液相色譜儀,以保留時間定性,測定峰面積,計算供試品中10種人參皂苷的含量。3種劑型供試品中人參皂苷含量RSD(n=6)均小于3%,結果表明方法重復性良好,結果見表3。
2.6 系統適應性考察 取10種人參皂苷混合對照品工作液10 μL進樣,計算10種人參皂苷的理論板數。得到人參皂苷Rg3、Rg1、Re、Rf、Rg2、Rb1、Rc、Rb2、Rb3、Rd的理論板數分別為103 427、50 732、104 490、157 284、120 457、82 876、253 440、260 991、410 628、239 554,分離度分別為5.4、1.6、32.6、15.1、2.2、4.0、4.8、1.6、8.0。對于供試品,雖然存在基質干擾影響分離度,但是3種劑型供試品中10種人參皂苷均能達到基線分離,分離度均能達到1.5以上。

表3 重復性試驗結果
注:“-”表示未檢出或低于定量限
2.7 精密度試驗 取10種人參皂苷混合對照品工作液10 μL連續進樣5次,以測得的峰面積響應值作評價標準,得到10種人參皂苷的RSD(n=5)均小于3.0%,表明在本方法儀器條件下,儀器精密度良好。
2.8 穩定性試驗 分別取供試品S01、S05、S08,按“2.3”項下方法操作,得到供試品溶液,室溫下放置24 h,分別在0、2、4、8、12、24 h取10 μL進樣,得到10種人參皂苷峰面積RSD(n=6)都在3.0%以內,表明供試品溶液在24 h內穩定。
2.9 回收率試驗 準確量取6份已知含量的供試品(S01)4.0 mL至分液漏斗中,分別精密加入人參皂苷對照品儲備液適量(對照品加入量與供試品中各人參皂苷含量之比為1∶1),以下按“2.3.2”項下方法操作,即可得到加標溶液。
準確稱取已知含量的供試品(S05)0.5 g,供試品(S08)1 g,各6份,分別精密加入人參皂苷對照品儲備液適量(對照品加入量與供試品中各人參皂苷含量之比為1∶1),置于100 mL錐形瓶中,以下按“2.3.1”項下方法操作,即可得到加標溶液。
取10 μL注入液相色譜儀,以保留時間定性,測定峰面積,得到10種人參皂苷的平均加樣回收率(n=6),RSD均小于4.0%,結果見表4。

表4 回收率結果
注:“-”表示未檢出或低于定量限
2.10 檢出限與定量限 S/N=3時,得到檢出限LOD,人參皂苷Rg1、Rg2、Rg3、Rb1、Rb2、Rb3、Rc、Rd、Re、Rf檢出限分別為0.002 4、0.002 1、0.002 9、0.001 9、0.001 8、0.002 1、0.002 2、0.002 1、0.002 1、0.002 0 μg;S/N=10時,得到定量限LOQ,定量限分別為0.006 0、0.005 4、0.007 4、0.005 0、0.004 4、0.005 3、0.005 6、0.005 2、0.005 2、0.005 0 μg。
2.11 供試品的測定 取12批供試品,按照按“2.3” 制備供試品溶液,每批平行處理2份,按上述色譜條件進行測定,將峰面積代入“2.4”線性回歸方程計算含量,結果見圖1~2及表5。

表5 供試品中10種成分含量測定結果
注:“-”表示未檢出或低于定量限
3 討論
3.1 前處理考察 由于保健食品劑型種類多,而每種劑型的基質比較復雜,導致10種人參皂苷更難同時分離。首先,通過比較3種不同的提取試劑,水飽和正丁醇、甲醇和乙醇,最終得到水飽和正丁醇提取效率最高。其次,選用水飽和正丁醇分別采用回流提取、液-液萃取、浸泡放置過夜超聲提取和直接超聲提取4種提取方式進行比較,結果表明:對于片劑和膠囊劑,浸泡過夜超聲提取與回流提取得到皂苷含量最高,又因為前者操作簡單,且提取的多糖等雜質較少,最終采用浸泡過夜超聲提取;對于口服溶液,回流提取與液-液萃取都能得到較高總皂苷含量,優先選取重現性好且操作較簡單的處理方法,因此采用水飽和正丁醇振搖多次萃取。
3.2 流動相及梯度的選擇 本文對甲醇-水,乙腈-水和乙腈-0.1%磷酸溶液3種不同流動相進行比較,結果表明,人參皂苷在低波長范圍內檢測時,乙腈比甲醇背景噪音低,可獲得較好的分離效果,并且乙腈與水混合黏度小,可以有效降低系統壓力,而加入磷酸對整體分離情況沒有明顯改善且磷酸鹽對色譜柱損耗大,最終選擇乙腈-水作為最佳流動相。
10種人參皂苷中Rg1和Re,Rb2和Rb3較難分離。人參皂苷Rg1和Re極性非常相似,較難分離,且供試品在人參皂苷Rg1和Re附近有雜質干擾,最終選擇合適梯度,在45 min 左右達到基線分離。Rb2和Rb3是同分異構體,并且兩者含量很低,容易包裹在雜質峰中,本試驗在保證峰形和柱效的前提下完成了兩種皂苷的基線分離。故最終采用梯度洗脫使每種皂苷達到較好分離效果。
3.3 樣品測定結果分析 由表5可見,12批供試品10種皂苷含量之和差異很大,含量最高的為硬膠囊,片劑和軟膠囊次之,口服溶液最低。每批供試品中,單種人參皂苷占10種皂苷比例各不相同,經過分析發現,Rb1、Rc、Rd、Re 4種所占比例最大,7批供試品含這4種皂苷比例為67.0%~88.5%,4批供試品的比例為39.0%~53.8%,1種供試品(S04)比例為0。對于供試品(S04),根據《保健食品檢驗與評價技術規范》(2003年版)中規定的紫外分光光度法進行總皂苷檢測,得到總皂苷含量為80 mg·100 mL-1。本文建立的HPLC-PAD法可對西洋參類保健食品中皂苷成分進行初步鑒定,最終用紫外分光光度法進行總皂苷檢測。
4 結論
本文共收集口服溶液、片劑和膠囊劑12批西洋參類保健食品,通過測定其線性范圍、系統適用性、重復性、精密度、穩定性、檢出限、定量限和回收率試驗,結果令人滿意。試驗表明,在本文供試品制備方法和色譜條件下,人參皂苷Rg3、Rg1、Re、Rf、Rg2、Rb1、Rc、Rb2、Rb3、Rd能夠達到完全分離,所建立的方法操作簡便,重復性好,可以用來對以西洋參為原料的保健食品進行質量控制。
