高效液相色譜法測定附子理中丸中6-姜辣素的含量
2020-06-24 14:04:44潘海霞王曉琦武紅娜
食品與藥品 2020年3期
潘海霞,王曉琦,武紅娜
(滄州市食品藥品檢驗所,河北 滄州 061000)
附子理中丸于2009年載入《國家基本藥物目錄》,是甲類處方藥,臨床應用廣泛。其處方出自宋代的《太平惠民和劑局方》,由附子(制)、黨參、炒白術、干姜、甘草5味藥組方而成[1-5]。干姜是附子理中丸方中君藥[5],6-姜辣素為其有效成分,具有抗胃潰瘍、止吐的藥理作用[6-8],與附子理中丸脾胃虛寒、嘔吐泄瀉的功能相對應,可在一定程度上反映該藥的功效。6-姜辣素性質不穩定,不同產地、不同采收季節干姜中6-姜辣素含量會有所變化,生產過程中容易造成6-姜辣素成分流失[9-11]。因此選擇6-姜辣素作為附子理中丸檢驗的新指標。《中國藥典》2015年版一部為該藥的法定標準,標準中僅對干姜進行了顯微鑒別和薄層色譜鑒別,而6-姜辣素并未進行監測,故本文擬定建立高效液相色譜法(HPLC)對6-姜辣素含量進行檢驗,以期控制附子理中丸的藥品質量。
1 儀器與試藥
1.1 儀器
Agilent 1200型高效液相色譜儀(美國安捷倫公司);CP225D型十萬分之一電子天平(德國賽多利斯公司);LC350A型超聲波處理器(魯超超聲設備有限公司);Milli-Q Advantage A10型超純水儀(美國Millipore公司)。
1.2 試藥
6-姜辣素對照品(批號:111833-201806,含量以99.9 %計,-20 ℃保存,中國食品藥品檢定研究院);甘草、白術、黨參對照藥材(批號分別為:120904-201620,120925-201812,121057-201206,供鑒別用),均購自中國食品藥品檢定研究院;附子對照藥材(批號為PCS8186,供鑒別用),購自北京中科質檢生物技術有限公司;附子理中丸來自不同廠家、3種劑型共計15批次;甲醇、乙腈均為色譜純,水為超純水(自制),其他試劑均為分析純。
2 方法與結果
2.1 色譜條件
色譜柱:Agilent 588935-902 TC-C18(2)(150 mm×4.6 mm,5 μm);流動相:甲醇-乙腈-水(30:25:45),等度洗脫;流速:0.8 ml/min;柱溫:25 ℃;檢測波長:280 nm;進樣量10 μl。理論板數按6-姜辣素計算不低于5000,色譜峰與其相鄰色譜峰的分離度均大于1.5。
2.2 溶液的制備
2.2.1 對照品溶液的制備 取6-姜辣素對照品適量,精密稱定,加甲醇制成每1 ml含0.1 mg的溶液,即得。
2.2.2 供試品溶液的制備 取本品水蜜丸適量,研細,取約2.2 g,精密稱定,或取小蜜丸或大蜜丸,剪碎,取約3.2 g,精密稱定;置具塞錐形瓶中,精密加入75 %乙醇20 ml,密塞,稱定重量,超聲處理(功率250 W,頻率30 kHz)25 min,放冷,用75 %乙醇補足減失的重量,搖勻,0.45 μm微孔濾膜過濾,即得。
2.2.3 陰性對照溶液的制備 按附子理中丸(濃縮丸)處方及工藝制備缺少干姜的陰性樣品,并按2.2.2項下供試品溶液制備方法制成陰性對照溶液。
2.3 方法學驗證
2.3.1 專屬性試驗 分別取對照品溶液、供試品溶液和陰性對照溶液,依據2.1項下色譜條件測定,結果見圖1。供試品溶液的色譜圖中,在與對照品溶液的色譜圖相應的位置上,有相同保留時間的色譜峰,而陰性對照溶液在此保留時間處無干擾。
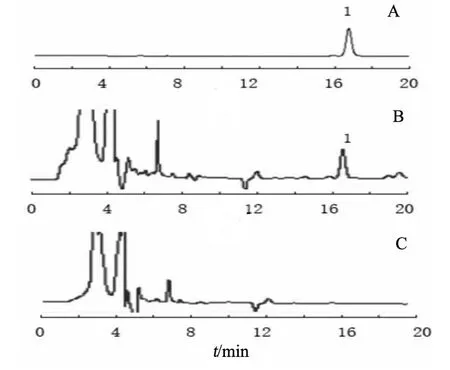
圖1 6-姜辣素的HPLC圖譜
2.3.2 線性關系考察 取6-姜辣素對照品適量,精密稱定,加適量甲醇制成濃度分別為0.04,0.06,0.10,0.20,0.30 mg/ml的溶液,分別精密吸取上述不同濃度的對照品溶液各10 μl,按照2.1項下色譜條件測定。以峰面積(y)對濃度(x,mg/ml)進行線性回歸,得回歸方程為:y=5706.5x+22.048,相關系數r=0.9986。結果表明,6-姜辣素在0.04~0.30 mg/ml范圍內與峰面積線性關系良好。
2.3.3 溶液穩定性 將供試品溶液(2號企業,批號為180101)分別于配置后在室溫下放置0,2,4,8,12和24 h,依法測定其峰面積,結果6個檢測點6-姜辣素的峰面積的RSD為0.4 %,說明供試品溶液在24 h內穩定。
2.3.4 精密度試驗 取2.2.1項下對照品溶液,按2.1項下色譜條件連續進樣6次,測得6-姜辣素峰面積的RSD為0.7 %,表明儀器精密度良好。
2.3.5 重復性試驗 取同批號樣品,按2.2.2項下方法平行制備6份供試品溶液(2號企業,批號為180101),按2.1項下色譜條件測定。結果,樣品中6-姜辣素平均含量為4.5 mg/丸,RSD為1.27 %(n=6),結果表明該方法的重復性良好。
2.3.6 回收率測定 取已知含量的樣品(2號企業,批號為180101)約3.2 g,精密稱定,分別按樣品含量的50 %,100 %,150 %加入6-姜辣素對照品,按2.2.2項下方法制備成供試品溶液,按2.1項下色譜條件測定,測得6-姜辣素的回收率范圍為97.75 %~102.58 %,平均回收率為99.43 %,RSD為1.5 %,結果表明加樣回收率良好。
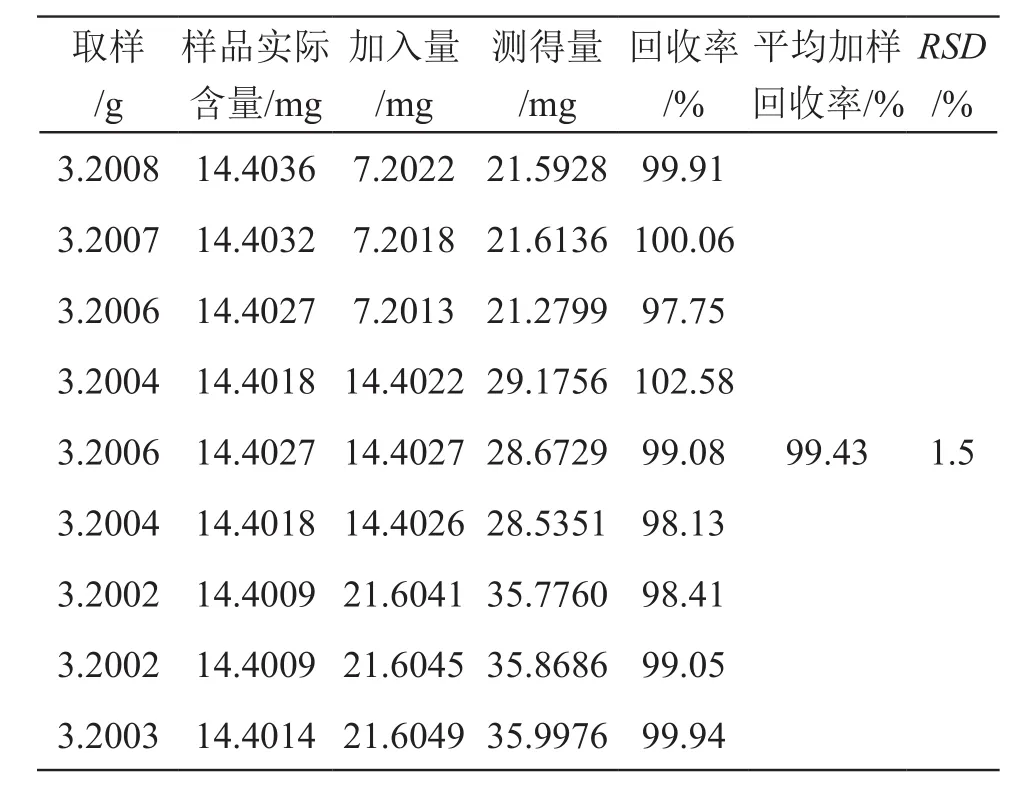
表1 回收率試驗結果
2.4 樣品測定
取15批樣品(大蜜丸、小蜜丸、水蜜丸),按2.2.2項下方法制備供試品溶液,按2.1項下色譜條件測定,計算6-姜辣素含量。結果見表2。由表2可見,3種劑型附子理中丸6-姜辣素含量差異較大,作為藥品的活性成分,亟待制定標準進行質量控制。
3 結論與討論
3.1 試驗結果分析
不同劑型、廠家樣品中6-姜辣素含量有明顯差異。分析原因:一是原藥材質量較差,不同產地、不同采收季節干姜中6-姜辣素含量會有所變化;二是工藝方面,水蜜丸在制丸干燥或滅菌過程中均有可能造成6-姜辣素成分流失。
3.2 提取溶劑的選擇
分別采用75 %乙醇、75 %甲醇、乙醇、甲醇、水進行提取,發現75 %甲醇提取較完全,且峰形較好,穩定性好,保留時間適中,考慮到實驗效率和效果及儀器的穩定狀態等綜合因素,確定使用75 %甲醇為溶劑。
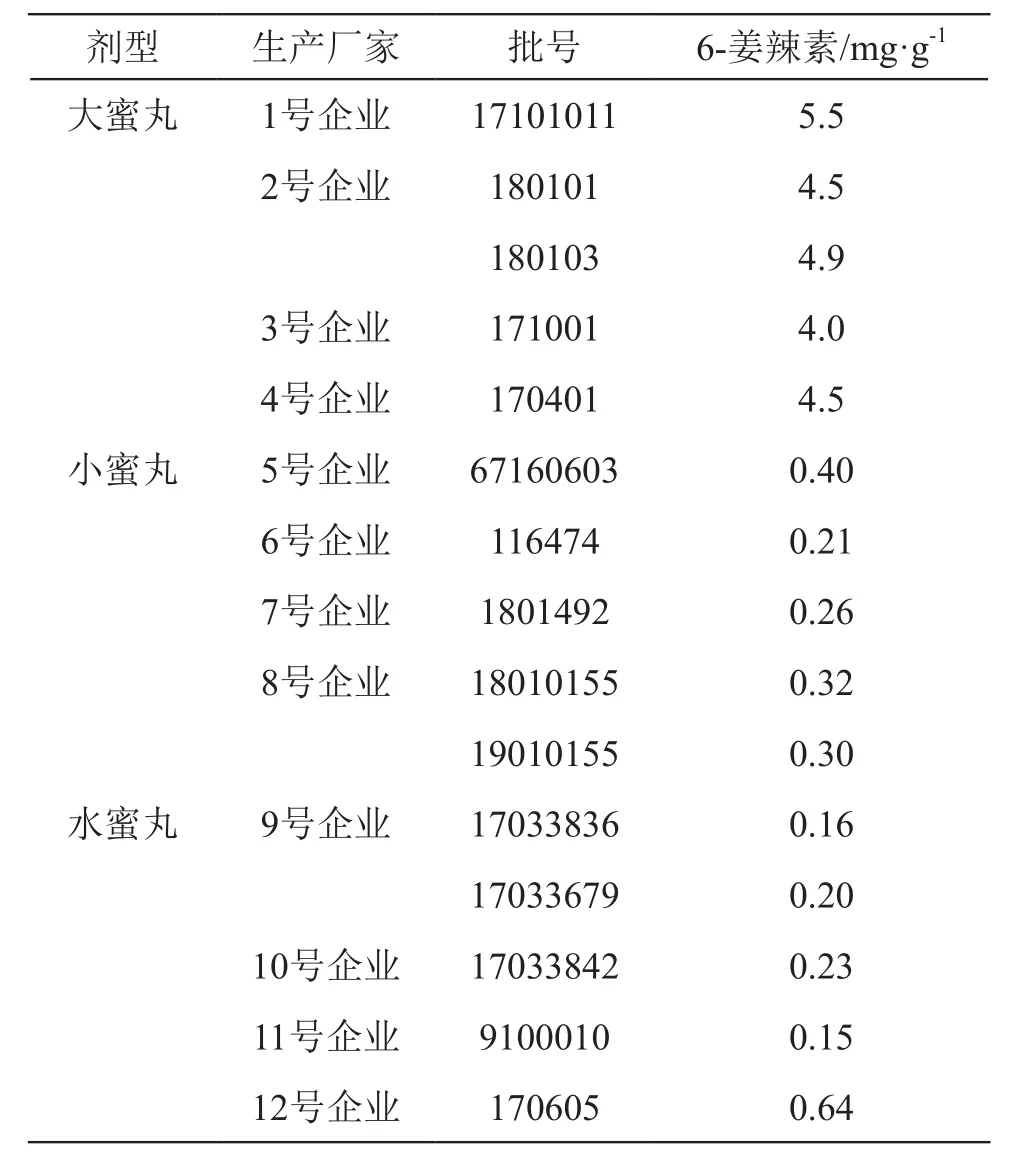
表2 樣品測定結果
3.3 提取方式的考察
分別采用了75 %甲醇溶解,超聲15,20,40 min,振搖50,40,30 min,放置24 h,發現超聲20 min至40 min與振搖30 min提取比較完全,但隨超聲時間的延長,6-姜辣素的含量會降低,綜合考慮到實驗效率,確定為超聲20 min。
3.4 柱溫的考察
針對在含量測定中,6-姜辣素作為干姜的活性成分,但其性質不穩定。實驗中考察了柱溫(20,25,30,35 ℃)對色譜分離的影響,結果表明隨柱溫升高,供試品溶液的峰形較差,分離效果也變差,而當柱溫在 25 ℃ 時,峰形較好,分離情況較好,故建議柱溫控制在25 ℃。
3.5 方法學考察
結果表明,本實驗方法操作簡便、專屬性強、重現性好,可準確測出附子理中丸中6-姜辣素的含量,因此可用于附子理中丸及其同類藥品中6-姜辣素含量的控制。
