頭花蓼質量標準的研究
2020-05-12 13:37:30趙英魁黃豆豆王璇江圣圭孫連娜
中成藥 2020年2期
趙英魁黃豆豆王 璇江圣圭孫連娜?
(1.海軍軍醫大學藥學院,上海 200433;2.上海中醫藥大學中藥學院,上海 201203;3.福建中醫藥大學藥學院,福建 福州 350108)
頭花蓼又名太陽草、石莽草、水繡球等,為蓼科蓼屬植物頭花蓼Polygonum capitatumBuch.-Ham.ex D.Don 的干燥全草或地上部分,是少數民族常用藥。在《廣西中藥志》 《貴州省中藥材、民族藥材質量標準》[1]《中國藥用植物志》 《云南中草藥》 《中藥大辭典》 《中華本草》[2]等書籍中均有記載。全草入藥,味苦、辛,性涼。頭花蓼含有黃酮類[3-4]、酚酸類[5-7]、木脂素類[8]等,具有抗菌[9-10]、抗炎鎮痛、抗氧化[11]、降糖[8]等藥理作用。主要用于清熱解毒、利尿通淋、腎盂腎炎、尿路結石、膀胱炎、風濕痛、跌打損傷、尿道感染、瘡瘍濕疹等癥。目前,頭花蓼被收載于2003 年版《貴州省中藥材、民族藥材質量標準》、2009 年版《湖南省中藥材標準》。在兩者地標中只規定了性狀、TLC 鑒別、水分檢查、含有量測定等項目,相關標準不夠全面,且含有量測定項中只以槲皮素為質量控制指標,供試品溶液制備了鹽酸水解方法。以頭花蓼為原料的單方制劑熱淋清顆粒[12]收載于2015 年版《中國藥典》 的含有量測定項以沒食子酸為對照品。課題組參考相關文獻[13-19],選擇代表性活性成分沒食子酸和槲皮苷,運用顯微鑒別法、TLC 法和HPLC 法對23 批藥材進行了定性鑒別和含有量測定,并對藥材的水分、總灰分、酸不溶性灰分等進行檢查,初步制定檢測限度,對其質量標準進行具體研究,以期為完善該藥材的質量控制提供依據。
1 材料
Waters 1525 高效液相色譜儀(美國沃特世公司);Sartorius CPA225D 電子分析天平(賽多利斯科學儀器北京有限公司);KQ-500 型超聲儀(昆山市超聲儀器有限公司);Biomate 3S UV-visible spectrophotometer(美國賽默飛世爾科技公司)。
正相硅膠HPTLC Silica gel 60(德國默克公司,貨號1.05641.0001,批號HX257461)。乙腈(色譜純)、水為蒸餾水、其他試劑均為分析純。沒食子酸對照品(批號110831-201204)、槲皮苷對照品(批號111538-200504)均購于中國食品藥品檢定研究院。
頭花蓼藥材對照品及23 批頭花蓼藥材,由海軍軍醫大學陳萬生教授鑒定為正品,信息見表1。
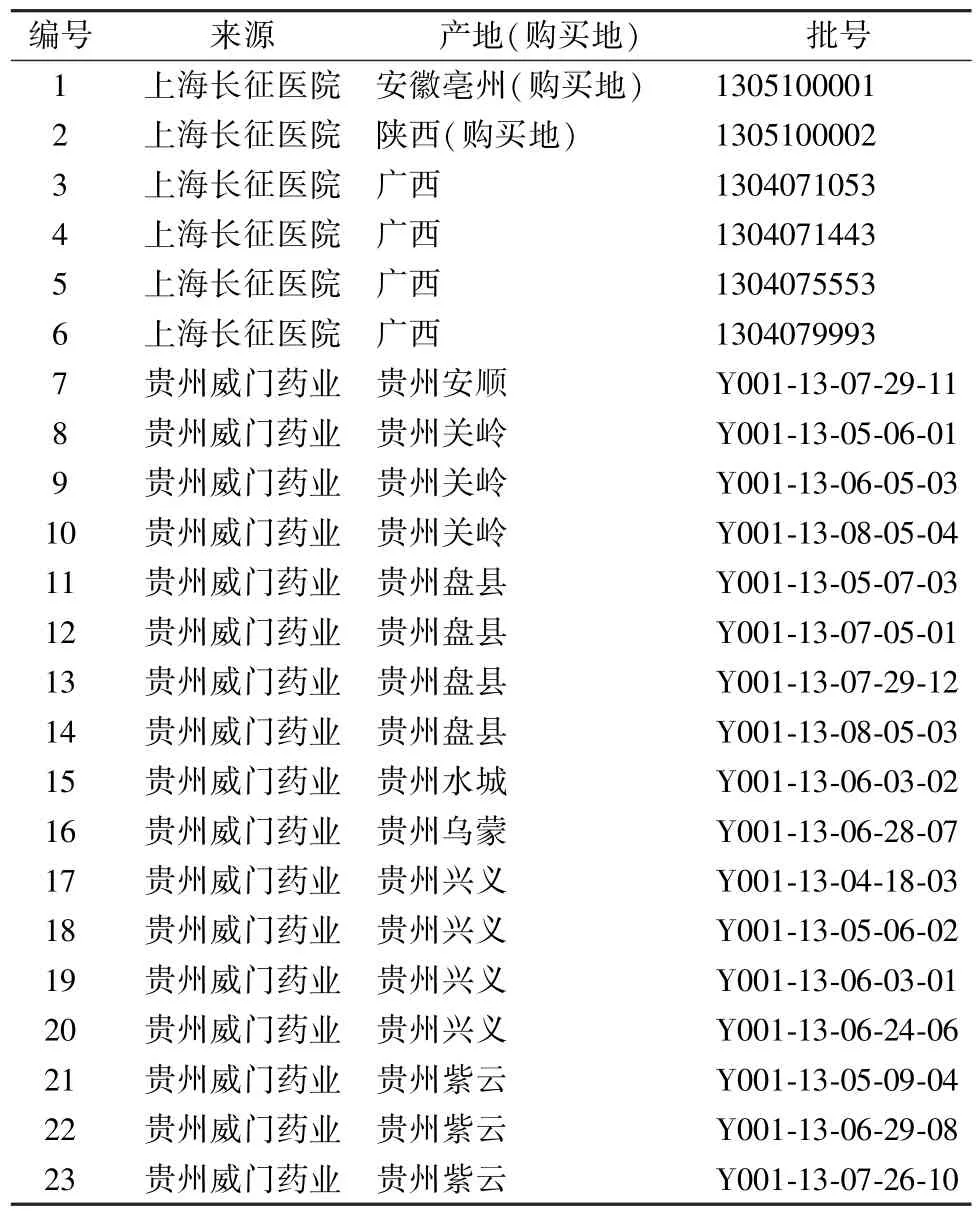
表1 樣品信息Tab.1 Information of samples
2 方法與結果
2.1 顯微鑒別 對頭花蓼藥材葉的表面觀(臨時水裝片)及藥材粉末(水合氯醛裝片)的相關顯微特征進行鑒別觀察。葉的表面觀,上表皮細胞垂周壁近平直,下表皮細胞垂周壁略呈波狀彎曲。氣孔不定式,副衛細胞3~4 個,上下表皮均有分布,下表皮較多。腺毛可見,常含有紅棕色物質,腺柄由1~3 列細胞組成,細胞壁明顯增厚,并具疣狀突起。粉末特征,本品粉末棕褐色,草酸鈣簇晶多,大小懸殊,直徑10~75 μm。纖維多成束或散在,易斷裂,多壁厚,具明顯孔溝。可見螺紋及環紋導管。淀粉粒眾多,常見單粒,類圓形、卵圓形等。顯微特征見圖1。

圖1 頭花蓼葉表面和藥材粉末顯微特征圖Fig.1 Micropic characteristics graph of leaf surface of P. capitatum and medicinal materials powder
2.2 TLC 定性鑒別
2.2.1 展開體系優化 考察二氯甲烷-乙酸乙酯-甲酸(2∶8∶1)、正己烷-乙酸乙酯-甲酸(2∶8∶0.5)、甲苯-乙酸乙酯-甲酸(2∶6∶1.5)、甲苯-乙酸乙酯-甲酸(2∶8∶1)、甲苯-乙酸乙酯-甲酸(2∶10∶1)、甲醇-水-甲酸(9∶5∶5)等展開體系,結果發現在甲苯-乙酸乙酯-甲酸(2∶8∶1)體系下,斑點清晰,分離度好。
2.2.2 載體考察 在甲苯-乙酸乙酯-甲酸(2∶8∶1)體系下,考察硅膠G 預制板、聚酰胺薄膜、高效硅膠G 預制板3 種載體,結果發現高效硅膠G預制板為載體時,斑點分離完全,較清晰,展開結果理想。
2.2.3 耐用性考察 考察不同品牌薄層板[銀龍牌的硅膠HSG 預制板(批號20130531)、黃海牌硅膠HSG 預制板(批號20130601)、默克正相硅膠HPTLC Silica gel 60(貨號1.05641.0001,批號HX257461)];不同溫度(4、12、22 ℃);不同濕度(32%、52%、72%),發現硅膠G 板、溫度、濕度對結果影響不大,展開結果均較好,表明該方法耐用性良好。
2.2.3 方法與結果 取頭花蓼粉末1 g,置50 mL具塞錐形瓶中,加20% 甲醇20 mL,超聲處理30 min,取出,濾過,濾液蒸干,殘渣用甲醇定容至1 mL 量瓶中,作為供試品溶液。精密稱取適量沒食子酸、槲皮苷對照品,加甲醇分別制成每1 mL含1.0、0.5 mg 溶液,作為對照品溶液。另取頭花蓼對照藥材1 g,同法制備對照藥材溶液。照TLC 2015 年版《中國藥典》 四部0502 試驗,吸取上述供試品和對照藥材溶液3 μL、對照品溶液各2 μL,分別點于同一高效硅膠G 薄層板上,以甲苯-乙酸乙酯-甲酸(2∶8∶1)為展開劑,展開,取出,晾干,噴以5%三氯化鋁乙醇溶液,熱風吹至斑點顯色清晰,置紫外光燈(365 nm)下檢視,結果見圖2。供試品色譜中,在與對照藥材和對照品色譜相應的位置上,顯相同顏色的熒光斑點,斑點清晰,分離度好。

圖2 頭花蓼TLC 圖Fig.2 TLC chromatograms of P. capitatum
2.3 水分、總灰分、酸不溶性灰分、浸出物檢查 根據2015 年版《中國藥典》 四部方法測定,結果見表2。
2.4 沒食子酸含有量測定
2.4.1 波長選擇 將沒食子酸對照品溶液,在200~700 nm 范圍內掃描,發現沒食子酸在220、273 nm 波長處有最大吸收,按《中國藥典》 有關規定,先檢查了所使用乙腈溶劑在供試品測定所用波長附近符合要求,結合其他相關文獻,最后選擇270 nm 作為沒食子酸的檢測波長。
2.4.2 流動相選擇 參照2015 年版《中國藥典》一部及相關文獻,廣棗、余甘子、藍布正等文獻資料中對沒食子酸含有量測定的條件,考察了甲醇-0.2%磷酸(18∶82)、乙腈-0.2% 甲酸(5∶95)、乙腈-0.2%磷酸(5∶95)對沒食子酸的洗脫效果,結果發現乙腈-0.2%磷酸(5∶95)流動相下,沒食子酸的峰型較好,與周邊峰無相互干擾,達到完全分離。故選擇選擇乙腈-0.2%磷酸(5∶95)作為流動相。
2.4.3 色譜條件 迪馬Dikma Diamonsil C18色譜柱(4.6 mm×250 mm,5 μm);流動相乙腈-0.2% 磷酸溶液(5∶95);檢測波長270 nm;體積流量1 mL/min;進樣量20 μL。
2.4.4 對照品溶液制備 精密稱取沒食子酸對照品5.52 mg,置10 mL 量瓶中,加甲醇溶解定容搖勻,得質量濃度為0.552 mg/mL 的對照品貯備液。
2.4.5 供試品溶液制備 取本品粉末(過3 號篩)約0.5 g,精密稱定,置50 mL 具塞錐形瓶中,精密加入10%甲醇25 mL,稱定質量,置水浴中加熱回流60 min,放冷,稱定質量,用10%甲醇補足減失質量,搖勻,濾過,取續濾液,即得。

表2 水分、總灰分、酸不溶性灰分、浸出物含有量測定結果Tab.2 Results of content determination of water,total ash,acid-insoluble ash and ectract
2.4.6 系統適用性試驗 在“2.4.1”項色譜條件下,分別取沒食子酸對照品溶液、供試品溶液注入液相色譜儀,沒食子酸的保留時間8.2 min,沒食子酸和其相近的其他峰分離完全(分離度>1.5),即本實驗條件下沒食子酸與其他組分分離完全。理論板數以沒食子酸計算為3 000,色譜圖見圖3。
2.4.7 線性關系考察 將沒食子酸對照品貯備液用甲醇 稀釋制成 552、276、138、69、34.5、17.25、8.626 μg/mL 的系列溶液,在“2.4.1”項色譜條件下測定,記錄峰面積,以峰面積為縱坐標(Y),沒食子酸質量為橫坐標(X)進行回歸,得回歸方程為Y=2 959 114.4X+67 276.7,r=0.999 9,表明沒食子酸在0.1725~11.04 g 范圍內線性關系良好。
2.4.8 精密度試驗 精密吸取沒食子酸對照品溶液20 μL,在“2.4.1”項色譜條件下,重復進樣6次,測得峰面積RSD 為0.92%,表明儀器精密度良好。
2.4.9 穩定性試驗 精密吸取1 號藥材供試品溶液20 μL,在“2.4.1”項色譜條件下,分別于0、4、8、12、24 h 進樣,測得峰面積RSD 為1.90%,表明供試品溶液在24 h 內穩定性良好。

圖3 沒食子酸HPLC 色譜圖Fig.3 HPLC chromatograms of gallic acid
2.4.10 重復性試驗 取1 號藥材,平行制備6 份供試品溶液,在“2.4.1”項色譜條件下,分別進樣20 μL,記錄峰面積。得沒食子酸平均含有量為1.614 1 mg/g,RSD 為1.67%,表明該方法重復性良好。
2.4.11 加樣回收率試驗 精密稱定1 號樣品(沒食子酸含有量1.614 1 mg/g)約0.25 g,共6 份,置于具塞錐形瓶中,精密加入樣品含有量100%的沒食子酸對照品,按“2.4.3”項下方法制備供試品溶液,在“2.4.1”項色譜條件下測定,得沒食子酸平均加樣回收率為98.90%,RSD 為1.90%,結果見表4。
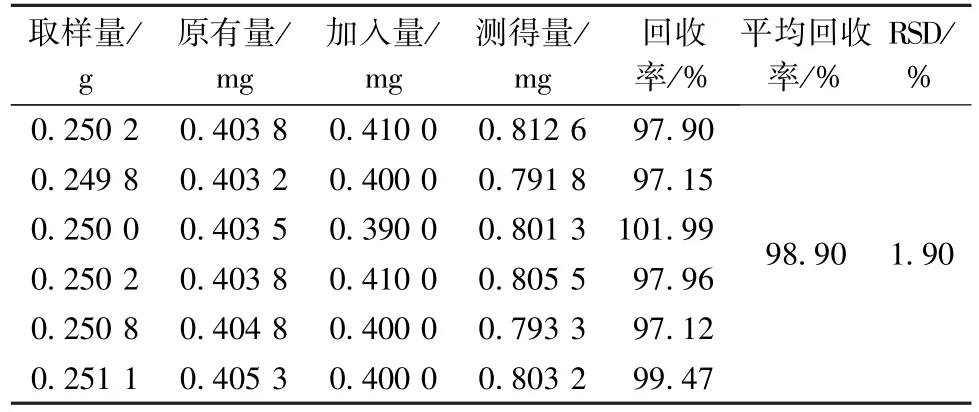
表4 沒食子酸加樣回收率試驗結果(n=6)Tab.4 Results of recovery tests for gallic acid(n=6)
2.4.12 樣品含有量測定 分別取上述23 批藥材,按“2.4.3”項下方 法制備 供試品溶液,在“2.4.1”項色譜條件下測定,結果見表5。
2.5 槲皮苷含有量測定
2.5.1 波長選擇 將槲皮苷對照品溶液,在200~700 nm 范圍內掃描,發現槲皮苷在257、348 nm波長處有最大吸收,按《中國藥典》 有關規定,先檢查了所使用的乙腈溶劑在供試品測定所用波長附近是符合要求的,結合其他相關文獻,最后選擇254 nm 分別作為槲皮苷的檢測波長。

表5 沒食子酸含有量測定結果(n=3,mg/g)Tab.5 Results of content determination of gallic acid(n=3,mg/g)
2.5.2 流動相選擇 參照2015 年版《中國藥典》一部及相關文獻,側柏葉、合歡花等文獻資料中對槲皮苷含有量測定的條件,考察了甲醇-0.2%磷酸(48∶ 52)、甲醇-0.2% 磷酸(46∶ 54)、乙 腈-0.2%甲酸(21∶79)、乙腈-0.2%磷酸(25∶75)、乙腈-0.2%磷酸(5∶95)對槲皮苷的洗脫效果,結果發現乙腈-0.2%磷酸(5∶95)流動相下,槲皮苷的峰型較好,與周邊峰無相互干擾,達到完全分離。故選擇乙腈-0.2% 磷酸(5∶95)作為流動相。
2.5.3 色譜條件 迪馬Dikma DiamonsiL C18色譜柱(4.6 mm×250 mm,5 μm);流動相乙腈-0.2%磷酸溶液(21∶79);檢測波長254 nm;體積流量1 mL/min;進樣量20 μL。
2.5.4 對照品溶液制備 精密稱取槲皮苷對照品6.26 mg,置10 mL 量瓶中,加甲醇溶解定容搖勻,即得質量濃度為0.626 mg/mL 的對照品貯備液。
2.5.5 供試品溶液制備 取本品粉末(過3 號篩)約0.5 g,精密稱定,置50 mL 具塞錐形瓶中,精密加入80%甲醇25 mL,稱定質量,置水浴中加熱回流60 min,放冷,稱定質量,用80%甲醇補足減失質量,搖勻,濾過,取續濾液,即得。
2.5.6 系統適用性試驗 在“2.5.1”色譜條件下,分別取槲皮苷對照品溶液、供試品溶液注入液相色譜儀,槲皮苷的保留時間14.3 min,槲皮苷和其相近的其他峰分離完全(分離度>1.5),即本實驗條件下槲皮苷與其他組分分離完全。理論板數以槲皮苷計算為3 000,色譜圖見圖4。

圖4 槲皮苷HPLC 色譜圖Fig.4 HPLC chromatograms of quercitin
2.5.7 線性關系考察 將沒槲皮苷對照品貯備液用甲醇 稀釋制成 626、313、156.5、78.25、39.125、19.562 5、9.781 25 μg/mL 的系列溶液,分別吸取20 μL 注入液相色譜儀,記錄色譜圖,以峰面積為縱坐標(Y),沒食子酸質量為橫坐標(X)進行回歸,得回歸方程為Y=2 537 136.6X+34 562.2,r=0.999 9,表明沒槲皮苷在0.195 6~12.52 g 范圍內線性關系良好。
2.5.8 精密度試驗 精密吸取槲皮苷對照品溶液20 μL,在“2.5.1”色譜條件下,重復進樣6 次,測得峰面積RSD 為0.86%,表明儀器精密度良好。
2.5.9 穩定性試驗 精密吸取1 號藥材供試品溶液20 μL,在“2.5.1”色譜條件下,分別于0、4、8、12、24 h 進樣,測得峰面積RSD 為1.24%,表明供試品溶液在24 h 內穩定性良好。
2.5.10 重復性試驗 取1 號藥材,平行制備6 份供試品溶液,在“2.5.1”色譜條件下,分別進樣20 μL,記錄峰面積。得槲皮苷均含有量為3.891 8 mg/g,RSD為1.50%,表明該方法重復性良好。
2.5.11 加樣回收率試驗 精密稱定1 號樣品(槲皮苷含有量3.891 8 mg/g)約0.25 g,共6 份,置于具塞錐形瓶中,精密加入樣品含有量100%的槲皮苷對照品,按“2.5.3”項下方法制備供試品溶液,在“2.5.1”色譜條件下進樣,得槲皮苷平均加樣回收率為98.89%,RSD 為3.05%,結果見表6。
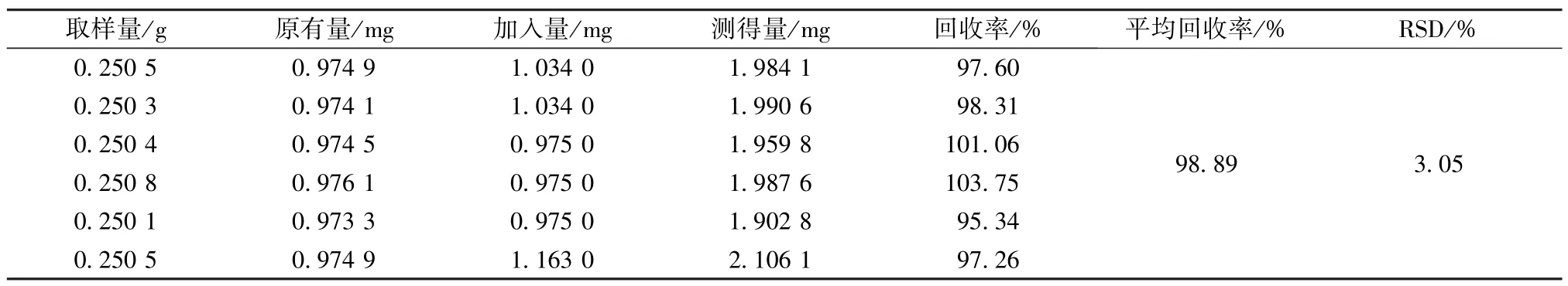
表6 槲皮苷加樣回收率試驗結果(n=6)Tab.6 Results of recovery tests for quercitin(n=6)
2.5.12 樣品含有量測定 分別取上述23 批藥材,按“2.5.3”項下方 法制備 供試品溶液,在“2.5.1”色譜條件下進樣,結果見表7。

表7 槲皮苷含有量測定結果(n=3,mg/g)Tab.7 Results of content determination of quercitin(n =3,mg/g)
3 結果與討論
3.1 檢查與浸出物限量 本實驗發現,23 批藥材水分范圍為7.75%~11.63%,平均為9.75%,暫定水分不得超過12.0%;總灰分范圍4.04%~18.79%,均值為11.47%,暫定總灰分不得超過14.0%;酸不溶性灰分范圍0.14%~8.62%,均值為2.62%,暫定酸不溶性灰分不得超過4.0%;50%乙醇浸出物范圍15.49%~25.26%,均值為20.50%,暫定不得低于15.0%。
3.2 沒食子酸、槲皮苷含有量和限量 實驗結果表明23 批樣品中沒食子酸、槲皮苷含有量差異較大,即使同一地區含有量也不盡相同。沒食子酸以陜西購的(2 號)含有量最高,貴州盤縣產地(14號)含有量最低;槲皮苷以貴州盤縣產地(13號)含有量最高,廣西產地(6 號)含有量最低。根據結果,暫定以干燥品計,沒食子酸含有量不得少于0.05%、槲皮苷含有量不得少于0.10%。
通過本研究,課題組完善了頭花蓼質量標準的研究,與貴州、湖南兩者地標相比,增加了顯微鑒別項,總灰分、酸不溶性灰分、50%乙醇浸出物檢查項,選擇代表性活性成分沒食子酸和槲皮苷作為質控指標,以期為制定頭花蓼質量標準提供依據,為完善藥材的質量控制控制奠定基礎。
