農(nóng)藥境外登記GLP項(xiàng)目的委托及審閱策略
2020-05-09 08:25:44李紅霞王文濤王欣童溫華珍林吉柏
世界農(nóng)藥 2020年4期
關(guān)鍵詞:報(bào)告實(shí)驗(yàn)室
李紅霞,高 靜,王文濤,王欣童,溫華珍,林吉柏
(北京穎泰嘉和分析技術(shù)有限公司,北京 102206)
我國是農(nóng)藥生產(chǎn)大國,也是農(nóng)藥出口大國。隨著農(nóng)藥市場(chǎng)的開放,我國農(nóng)藥企業(yè)加快開拓國外市場(chǎng)。農(nóng)藥進(jìn)入相關(guān)國家市場(chǎng)前須在各國相關(guān)部門進(jìn)行登記,獲取批準(zhǔn)。農(nóng)藥境外登記人員在準(zhǔn)備登記一個(gè)農(nóng)藥產(chǎn)品之前,需要先對(duì)該產(chǎn)品的可行性、運(yùn)營模式和登記時(shí)間、費(fèi)用等進(jìn)行分析,確定其數(shù)據(jù)缺口,從而進(jìn)行相關(guān)的試驗(yàn)安排。在獲取試驗(yàn)報(bào)告后,準(zhǔn)備相應(yīng)的卷宗,提交后跟蹤進(jìn)度,直至獲得登記許可。
良好實(shí)驗(yàn)室規(guī)范(Good Laboratory Practice,GLP)是一種質(zhì)量管理體系,是實(shí)驗(yàn)室研究從計(jì)劃、試驗(yàn)、監(jiān)督、記錄到試驗(yàn)報(bào)告等一系列的管理而制定的法規(guī)性文件,用于對(duì)農(nóng)藥、醫(yī)藥等化學(xué)品進(jìn)行安全性評(píng)價(jià)。GLP的目的就是降低試驗(yàn)的誤差,確保試驗(yàn)結(jié)果的真實(shí)性。許多國家在登記資料要求中,都規(guī)定企業(yè)提交的數(shù)據(jù)報(bào)告需由具有GLP資質(zhì)的實(shí)驗(yàn)室出具。因此,作為境外登記人員,需要了解GLP項(xiàng)目的基本程序,委托、跟蹤 GLP項(xiàng)目執(zhí)行及審閱GLP報(bào)告時(shí)應(yīng)重點(diǎn)關(guān)注的問題。本文概要介紹了境外登記人員如何進(jìn)行相關(guān)的試驗(yàn)委托,并對(duì)幾種常見的登記試驗(yàn)項(xiàng)目如何進(jìn)行審閱做了重點(diǎn)闡述。
1 選擇適合的GLP實(shí)驗(yàn)室
境外登記人員在進(jìn)行登記試驗(yàn)準(zhǔn)備工作時(shí)首先要確認(rèn)登記試驗(yàn)的種類,研究擬申請(qǐng)登記國家的相關(guān)要求,對(duì)可以提交查詢數(shù)據(jù)的材料,不需要安排GLP試驗(yàn),以避免不必要的資源浪費(fèi);但對(duì)登記資料要求中明確規(guī)定有關(guān)數(shù)據(jù)和報(bào)告必須符合GLP要求的項(xiàng)目,則必須選擇由具有GLP資質(zhì)的實(shí)驗(yàn)室開展試驗(yàn)并出具報(bào)告。此時(shí),選擇、確定合適的GLP實(shí)驗(yàn)室,就成為保障境外登記成功非常重要的一個(gè)環(huán)節(jié)。首先,境外登記人員要對(duì)備選的實(shí)驗(yàn)室資源進(jìn)行分析,考察GLP實(shí)驗(yàn)室的資質(zhì)、能力以及GLP報(bào)告提交該國的通過情況等;其次,對(duì)GLP實(shí)驗(yàn)室的報(bào)價(jià)、時(shí)間安排和周期等進(jìn)行考察。此外,境外登記人員還要特別注意備選的GLP實(shí)驗(yàn)室是否有多個(gè)地址,試驗(yàn)在哪里進(jìn)行等問題。
需要特別指出的一個(gè)細(xì)節(jié)是,境外登記人員在選擇GLP實(shí)驗(yàn)室時(shí)還要考慮到擬登記產(chǎn)品的性質(zhì)及運(yùn)輸條件,例如烯草酮這類易降解產(chǎn)品對(duì)運(yùn)輸條件要求較高,優(yōu)先選擇本地的GLP實(shí)驗(yàn)室會(huì)減少GLP試驗(yàn)的風(fēng)險(xiǎn)。
2 明確委托方及受托 GLP實(shí)驗(yàn)室的責(zé)任與義務(wù)
根據(jù)經(jīng)濟(jì)合作與發(fā)展組織(OECD) GLP準(zhǔn)則[1]的定義,委托方是指委托、資助及提交非臨床健康和環(huán)境安全研究的實(shí)體和單位。試驗(yàn)委托方在GLP準(zhǔn)則實(shí)施中有很多的任務(wù)和責(zé)任,GLP項(xiàng)目不單是GLP實(shí)驗(yàn)室的責(zé)任,委托方參與整個(gè)GLP項(xiàng)目的過程,從確定項(xiàng)目內(nèi)容,提供相應(yīng)信息開始,到樣品確認(rèn),方法篩選,雜質(zhì)確認(rèn),簽署研究計(jì)劃,試驗(yàn)期間的溝通,審閱、審批最終報(bào)告等,委托方都不可或缺。
委托方在項(xiàng)目運(yùn)行的各個(gè)階段都需要明確其相應(yīng)的職責(zé)并發(fā)揮作用:⑴ 試驗(yàn)前階段:委托方需要對(duì)實(shí)驗(yàn)室的資源進(jìn)行分析,確認(rèn)要進(jìn)行的試驗(yàn)類型,登記地所要求的試驗(yàn)指南,提供合適的被試物信息。明確委托方和試驗(yàn)機(jī)構(gòu)的各自職責(zé)。⑵ 計(jì)劃書階段:對(duì)預(yù)試驗(yàn)結(jié)果、試驗(yàn)計(jì)劃、企標(biāo)情況和可能出現(xiàn)問題等進(jìn)行溝通。⑶ 試驗(yàn)階段:當(dāng)試驗(yàn)結(jié)果出現(xiàn)問題或可疑數(shù)據(jù)等要及時(shí)溝通。例如動(dòng)物死亡的原因,劑量調(diào)整,異常數(shù)據(jù)等。⑷ 報(bào)告階段:要仔細(xì)審核草稿報(bào)告,對(duì)出現(xiàn)的問題及時(shí)聯(lián)系實(shí)驗(yàn)室進(jìn)行修改或者補(bǔ)充試驗(yàn)。當(dāng)提交最終報(bào)告遇到登記當(dāng)局的疑問時(shí),要及時(shí)聯(lián)系實(shí)驗(yàn)室協(xié)助解決。
在GLP項(xiàng)目的運(yùn)行中,委托方要注意把關(guān)好被試物的質(zhì)量,提供的相應(yīng)信息準(zhǔn)確無誤,并在GLP項(xiàng)目運(yùn)行期間和實(shí)驗(yàn)室保持良好溝通。與此同時(shí),承擔(dān)項(xiàng)目的GLP實(shí)驗(yàn)室要嚴(yán)格按照程序要求進(jìn)行試驗(yàn),確保試驗(yàn)的質(zhì)量,并及時(shí)匯報(bào)溝通。只有委托方和實(shí)驗(yàn)室雙方通力合作、溝通順暢,才能確保GLP項(xiàng)目的順利完成。
3 實(shí)施GLP項(xiàng)目的基本程序
進(jìn)行一個(gè)GLP項(xiàng)目的基本程序如下:⑴ 委托方和 GLP實(shí)驗(yàn)室簽署協(xié)議,確認(rèn)要進(jìn)行的項(xiàng)目及檢測(cè)內(nèi)容。⑵ 委托方提供被試物及相關(guān)信息。⑶ GLP實(shí)驗(yàn)室獲得樣品后展開預(yù)試驗(yàn)。⑷ GLP實(shí)驗(yàn)室撰寫項(xiàng)目試驗(yàn)計(jì)劃方案,質(zhì)量保證人員(QA)審核,委托方進(jìn)行確認(rèn)。⑸ GLP實(shí)驗(yàn)室根據(jù)審核批準(zhǔn)后的試驗(yàn)計(jì)劃實(shí)施GLP試驗(yàn)。⑹ GLP實(shí)驗(yàn)室根據(jù)試驗(yàn)結(jié)果出具草稿報(bào)告,經(jīng)QA和委托方審核后出具最終簽字版報(bào)告。
4 跟進(jìn)GLP項(xiàng)目試驗(yàn)過程
根據(jù)OECD GLP準(zhǔn)則[1]的要求,試驗(yàn)項(xiàng)目負(fù)責(zé)人(SD)是對(duì)試驗(yàn)項(xiàng)目的實(shí)施和管理全面負(fù)責(zé)的人員,SD的首要職責(zé)是確保研究工作遵從GLP的要求。在GLP試驗(yàn)體系中,質(zhì)量保證是一個(gè)獨(dú)立于試驗(yàn)操作、旨在保證試驗(yàn)單位遵循GLP準(zhǔn)則的系統(tǒng)。QA會(huì)科學(xué)、客觀地對(duì)試驗(yàn)設(shè)施、GLP軟硬件運(yùn)轉(zhuǎn)狀態(tài)、研究計(jì)劃、試驗(yàn)操作、原始數(shù)據(jù)及試驗(yàn)報(bào)告等是否符合GLP規(guī)范進(jìn)行監(jiān)督檢查。
GLP實(shí)驗(yàn)室對(duì)其GLP項(xiàng)目的試驗(yàn)過程有嚴(yán)格的要求,所有操作都需要按照實(shí)驗(yàn)室的標(biāo)準(zhǔn)操作程序進(jìn)行,試驗(yàn)過程嚴(yán)格按照計(jì)劃書執(zhí)行。計(jì)劃書、試驗(yàn)關(guān)鍵過程及最終報(bào)告QA都會(huì)進(jìn)行審查以確保研究工作遵從GLP。境外登記人員在GLP項(xiàng)目實(shí)施過程要和SD保持良好的溝通以便GLP項(xiàng)目順利實(shí)施。
GLP項(xiàng)目的最終成果是GLP報(bào)告,境外登記人員最需要關(guān)注的就是GLP項(xiàng)目的報(bào)告。根據(jù)GLP準(zhǔn)則要求,每個(gè)試驗(yàn)項(xiàng)目均應(yīng)當(dāng)有一份最終試驗(yàn)報(bào)告。試驗(yàn)項(xiàng)目負(fù)責(zé)人應(yīng)在最終試驗(yàn)報(bào)告上簽署姓名和日期,對(duì)數(shù)據(jù)有效性、真實(shí)性和完整性負(fù)責(zé),同時(shí)說明遵從本規(guī)范和試驗(yàn)計(jì)劃的程度,以及偏離對(duì)試驗(yàn)結(jié)果的影響。試驗(yàn)報(bào)告修訂應(yīng)明確說明修改或補(bǔ)充的理由,并由試驗(yàn)項(xiàng)目負(fù)責(zé)人簽署姓名和日期。此外,最終試驗(yàn)報(bào)告需要包括以下內(nèi)容:⑴ 試驗(yàn)項(xiàng)目基本情況;⑵ 委托方和試驗(yàn)單位的情況;⑶ 試驗(yàn)開始時(shí)間和完成時(shí)間;⑷ QA聲明,QA聲明中需要列出檢查類型、檢查內(nèi)容、檢查結(jié)果等信息;⑸ 儀器、試劑、材料和方法等信息;⑹ 項(xiàng)目中的偏離和修訂,需要說明偏離規(guī)范和試驗(yàn)計(jì)劃的程度,評(píng)價(jià)偏離的影響;⑺ 所做試驗(yàn)的描述和結(jié)果,需要報(bào)告所用試驗(yàn)計(jì)劃中要求的所有信息和數(shù)據(jù)。
境外登記人員在審閱GLP報(bào)告時(shí),尤其要注意報(bào)告中的GLP遵循部分的說明,這部分內(nèi)容會(huì)說明該項(xiàng)目對(duì)遵循GLP準(zhǔn)則程度或情況。正常情況下,GLP遵循中除被試物的信息或有些基于SD經(jīng)驗(yàn)做出的譜圖解析或解釋之外,其他不遵循GLP準(zhǔn)則的地方要特別引起重視,可能會(huì)對(duì)登記造成不良影響。此外,審閱人也要特別注意報(bào)告中的偏離和修訂,判斷其是否影響整個(gè)報(bào)告質(zhì)量。
5 審閱GLP報(bào)告及重點(diǎn)注意事項(xiàng)
GLP項(xiàng)目需要完全按照簽署的研究計(jì)劃進(jìn)行實(shí)施,而GLP報(bào)告需要完全精確地反映研究的實(shí)施和研究數(shù)據(jù)。不管是試驗(yàn)方案還是報(bào)告草案都需要仔細(xì)的審查,一旦簽字確認(rèn)后,根據(jù)GLP的規(guī)范,任何的修改都需要以修訂或偏離的形式出現(xiàn),并顯示在報(bào)告中。如果修訂或偏離過多,可能會(huì)降低登記審查機(jī)關(guān)對(duì)該GLP報(bào)告的好感,影響登記進(jìn)程。無論是GLP實(shí)驗(yàn)室,還是作為委托方代表的登記人員或咨詢顧問,都要在草案階段盡可能的去避免相應(yīng)的錯(cuò)誤和疏漏。
中國絕大多數(shù)企業(yè)的產(chǎn)品境外登記是相同產(chǎn)品登記,本文就目前市場(chǎng)相同產(chǎn)品登記中經(jīng)常使用的幾種類型的GLP項(xiàng)目,整理了一些需要重點(diǎn)注意的事項(xiàng)以供參考。
5.1 GLP項(xiàng)目中的信息審閱
根據(jù)不同國家的要求,檢測(cè)項(xiàng)目和導(dǎo)則會(huì)不同。在GLP項(xiàng)目的草稿計(jì)劃過程中,實(shí)驗(yàn)室和委托方一定要審查研究計(jì)劃中的檢測(cè)項(xiàng)目和對(duì)應(yīng)的試驗(yàn)方法和導(dǎo)則。提供準(zhǔn)確的樣品信息是委托方的責(zé)任,樣品信息雖然看起來簡單,但是出現(xiàn)的問題很多,要給予特別重視。樣品產(chǎn)品分析單(COA)中容易出現(xiàn)如下情況:⑴ COA中樣品名稱、物理特性等與GLP試驗(yàn)中使用的不一致;⑵ COA中質(zhì)檢報(bào)告日期晚于試驗(yàn)開始日期;⑶ COA中含量、批號(hào)與試驗(yàn)使用的不一致;⑷ 同一個(gè)批號(hào)供試品的COA不一樣,或者單位印章位置不同;⑸ 樣品COA信息與郵件、方案中不一致;⑹ 樣品COA保存條件前后不一致;⑺ 樣品COA中含量與GLP報(bào)告中差距較大;⑻ 樣品COA中只有公司印章,沒有責(zé)任人簽字;⑼ 需要注意,在巴西除五批次外的GLP報(bào)告都需要GLP的COA。
此外,在進(jìn)行GLP項(xiàng)目中,有時(shí)需要委托方提供其他與試驗(yàn)相關(guān)的數(shù)據(jù),如分散性、持久起泡性和懸浮性等,需要提供稀釋倍數(shù)等。
5.2 五批次項(xiàng)目審閱
農(nóng)藥原藥的五批次分析是指針對(duì)有效成分、含量達(dá)到0.1%及以上的任何顯著雜質(zhì),以及FAO (聯(lián)合國糧食及農(nóng)業(yè)組織)、WHO (世界衛(wèi)生組織)、歐盟或者是各國農(nóng)藥主管部門規(guī)定的相關(guān)雜質(zhì)定性和定量分析。五批次全分析鑒定出的組分總量在98%~102%,一般是由委托方提供五個(gè)批次的工業(yè)化原藥來進(jìn)行。
GLP的五批次報(bào)告是進(jìn)行國外市場(chǎng)開發(fā)及農(nóng)藥登記不可或缺的重要技術(shù)資料。目前絕大多數(shù)國家的登記都需要提供五批次報(bào)告,一些中高端市場(chǎng)更是日趨嚴(yán)格,越來越多的市場(chǎng)要求登記申請(qǐng)人提供 GLP五批次報(bào)告。五批次報(bào)告是解析原藥產(chǎn)品組分的核心登記資料,是判定原藥產(chǎn)品質(zhì)量是否合格的關(guān)鍵要素,也是農(nóng)藥藥效、毒理和環(huán)境等一系列試驗(yàn)的源頭。
GLP的五批次報(bào)告對(duì)判斷產(chǎn)品的質(zhì)量至關(guān)重要,該報(bào)告的結(jié)論直接反映了原藥的品質(zhì)。在幾乎所有的高端市場(chǎng),原藥的五批次分析報(bào)告還作為評(píng)價(jià)原藥產(chǎn)品是否符合相同產(chǎn)品的判定依據(jù),直接決定產(chǎn)品是否可以按照相同產(chǎn)品原則進(jìn)行登記——即是否可以以最低的登記成本取得登記。一份高質(zhì)量的五批次報(bào)告,幾乎可以在全球通用,可以說是市場(chǎng)開發(fā)的敲門磚。
對(duì)于GLP的五批次報(bào)告的具體要求,各國之間存在一定的差別。對(duì)于GLP的五批次報(bào)告的格式,除了符合OECD GLP導(dǎo)則中的規(guī)定內(nèi)容外,美國環(huán)境保護(hù)署(EPA)提出了一定的要求,如報(bào)告要增加是否有保密部分的聲明等[2]。按照美國EPA要求出具的GLP報(bào)告在其他國家提交基本沒有問題,一般不需要為各國準(zhǔn)備各自的報(bào)告模板。在五批次項(xiàng)目運(yùn)行過程中,實(shí)驗(yàn)室和委托方還要注意以下這些可能存在的問題。
5.2.1 各國對(duì)五批次的不同要求
一般而言,各國對(duì)五批次的定性、定量的要求基本一致,有效成分的定性一般2種方法就可以(一般接受核磁共振波譜和質(zhì)譜法定性),定量分析方法驗(yàn)證也基本都認(rèn)可遵從歐盟的SANCO 3030[3]指南。但在細(xì)節(jié)要求上,還存在一定的差異,需要實(shí)驗(yàn)室和委托方給予重視。
歐盟會(huì)根據(jù)生產(chǎn)工藝的資料來評(píng)估原藥整個(gè)生產(chǎn)過程中用到的所有溶劑或溶劑催化劑的毒性。毒性評(píng)估可以直接從官網(wǎng)上下載 MSDS (物質(zhì)安全數(shù)據(jù)單),并對(duì)毒性進(jìn)行比較。如果所用溶劑的毒性大于原藥主成分的毒性,就需要對(duì)溶劑進(jìn)行測(cè)定。在實(shí)驗(yàn)室檢測(cè)過程中,建議將定量限設(shè)為至多 0.01%或0.02%,以保證溶劑不會(huì)影響原藥的毒性。此外,溶劑測(cè)定的期限最好是距離生產(chǎn)日期近一些,這樣更容易反映出在生產(chǎn)過程中溶劑的真實(shí)殘留情況。
5.2.2 五批次GLP報(bào)告中的雜質(zhì)確認(rèn)及毒性評(píng)價(jià)
GLP的五批次分析一般包含2個(gè)階段,首先是實(shí)驗(yàn)室進(jìn)行的預(yù)掃描階段,這部分內(nèi)容不體現(xiàn)在GLP五批次報(bào)告中。在這個(gè)階段,實(shí)驗(yàn)室和委托方要確認(rèn)正式GLP試驗(yàn)中要進(jìn)行檢測(cè)的雜質(zhì),包括毒性相關(guān)雜質(zhì)和大于0.1%的任何顯著雜質(zhì)。這是一個(gè)五批次報(bào)告是否能順利拿到等同登記的關(guān)鍵,需要委托方和實(shí)驗(yàn)室共同確認(rèn)。在確認(rèn)好雜質(zhì)種類及準(zhǔn)備好雜質(zhì)標(biāo)準(zhǔn)品后,實(shí)驗(yàn)室就進(jìn)入到GLP階段,對(duì)有效成分及雜質(zhì)進(jìn)行定性、定量分析。
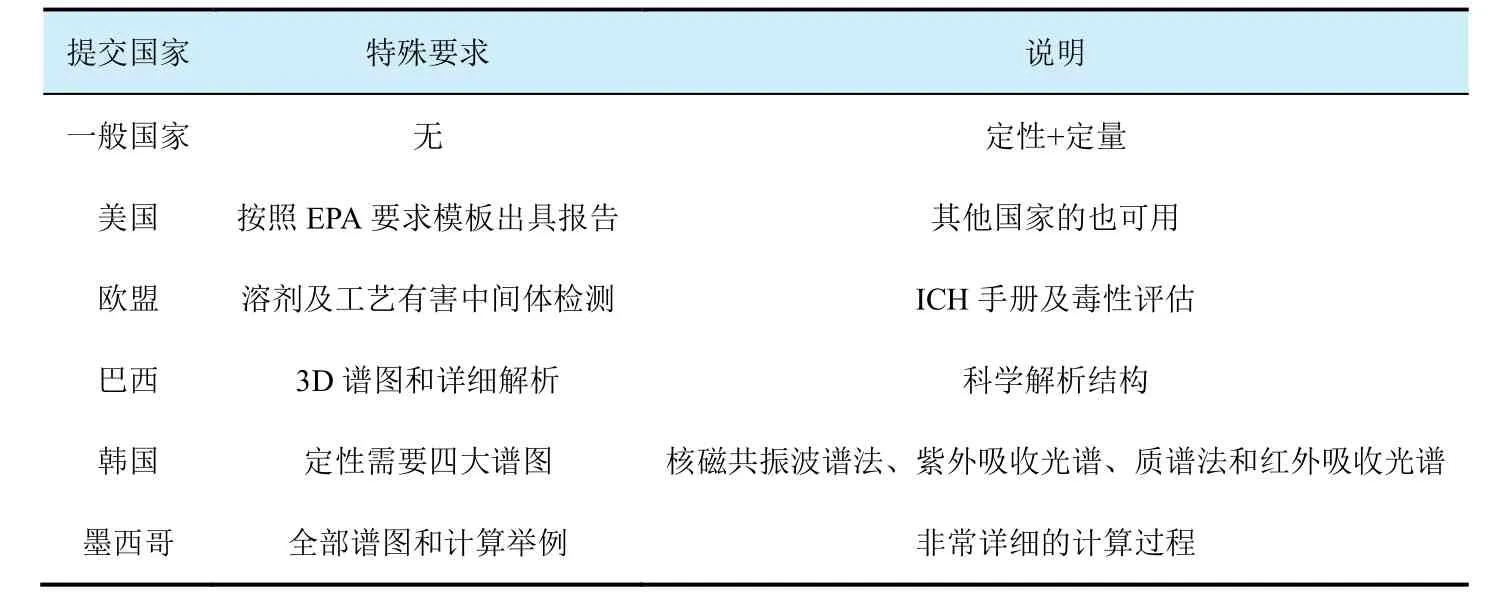
表1 各國對(duì)GLP五批次報(bào)告的特殊要求
因此,確認(rèn)及收集有關(guān)產(chǎn)品的雜質(zhì)信息非常重要,這對(duì)評(píng)估產(chǎn)品是否符合登記要求以及制定研究計(jì)劃等非常有幫助。除了FAO/WHO規(guī)格上會(huì)提到相關(guān)雜質(zhì),在歐盟、澳大利亞和加拿大等官網(wǎng)上可以查詢及下載已登記產(chǎn)品的評(píng)價(jià)報(bào)告,會(huì)提及需要關(guān)注的雜質(zhì)。GLP的五批次報(bào)告必須滿足登記國的要求,要考慮到報(bào)告的使用范圍,確認(rèn)好相關(guān)雜質(zhì)。
對(duì)于顯著雜質(zhì)而言,其是否會(huì)有毒性,是否會(huì)影響登記是委托方非常關(guān)心的問題。對(duì)于不確認(rèn)的雜質(zhì),可以根據(jù)預(yù)分析的結(jié)構(gòu),先用定量構(gòu)效關(guān)系(QSAR)來評(píng)判一下該化合物的毒性。QSAR是指定量的構(gòu)效關(guān)系,是使用數(shù)學(xué)模型來描述分子結(jié)構(gòu)和分子的某種生物活性之間的關(guān)系。有很多免費(fèi)的QSAR軟件可以幫助來進(jìn)行毒性評(píng)估,如 QSAR toolbox、Toxtree、VEGA等。
5.2.3 農(nóng)藥的儲(chǔ)存條件
大部分農(nóng)藥在常溫下儲(chǔ)存,其雜質(zhì)及性質(zhì)長時(shí)間不會(huì)發(fā)生改變。但有些特殊農(nóng)藥要特別注意特殊儲(chǔ)存條件以免影響到五批次的結(jié)果。吸水性強(qiáng)的農(nóng)藥,如煙嘧磺隆,需要在干燥條件下儲(chǔ)存,實(shí)驗(yàn)室在稱量樣品時(shí)也需要注意空氣中水分的影響。有些農(nóng)藥在常溫下易分解,主成分含量還沒有顯著變化時(shí),雜質(zhì)變化就已經(jīng)異常明顯,如烯草酮原藥,建議最好選擇國內(nèi)實(shí)驗(yàn)室進(jìn)行分析,且以冷凍狀態(tài)進(jìn)行運(yùn)輸,避免運(yùn)輸途中雜質(zhì)發(fā)生變化。
5.2.4 水分、不溶物等其他組分
水分含量是五批次項(xiàng)目中必須測(cè)試的項(xiàng)目。有些時(shí)候,根據(jù)不同產(chǎn)品的生產(chǎn)工藝,需要做干燥失重、不溶物或硫酸鹽灰分等項(xiàng)目。在進(jìn)行這些項(xiàng)目時(shí),要多思考其相關(guān)性。如干燥失重量遠(yuǎn)大于水分含量,需要解釋其他組分可能會(huì)是什么。五批次報(bào)告中的不溶物如果含量超過0.1%,在韓國登記時(shí)會(huì)被要求對(duì)這些不溶物進(jìn)行定性。
此外,在GLP五批次分析中,也要格外注意樣品的COA和五批次檢測(cè)項(xiàng)的符合程度,雖然可能由于測(cè)量方法的不同造成 COA中的含量和五批次結(jié)果中不一致,但是如果相差太大,將難以解釋。
5.2.5 農(nóng)藥的異構(gòu)體現(xiàn)象
農(nóng)藥的異構(gòu)體現(xiàn)象很普遍,其異構(gòu)體分幾何異構(gòu)體和光學(xué)異構(gòu)體。有的異構(gòu)體之間,生物活性有明顯甚至很大的差別,有的則差別不明顯。在GLP五批次報(bào)告中,實(shí)驗(yàn)室首先需要仔細(xì)查詢?cè)撧r(nóng)藥的有效組分究竟是其中的單一組分還是混合體。一般而言,如果有效成分是混合體,可以不用分開檢測(cè),但是各國對(duì)異構(gòu)體拆分要求的嚴(yán)格程度不同,需要根據(jù)登記市場(chǎng)進(jìn)行確認(rèn)。此外,雜質(zhì)的光學(xué)異構(gòu)體一般不再要求拆分檢測(cè)。
5.2.6 質(zhì)量平衡及產(chǎn)品規(guī)格
五批次GLP全分析分析報(bào)告中鑒定出的組分總量應(yīng)該為 98%~102%,當(dāng)測(cè)得的各種組分含量的總和不足98%時(shí),應(yīng)采用其他可能的方法進(jìn)一步鑒定分析;當(dāng)各種組分含量的總和超過102%時(shí),應(yīng)分析原因并做出合理解釋。絕大部分國家在制定登記產(chǎn)品的規(guī)格時(shí),是根據(jù)五批次GLP報(bào)告中的平均數(shù)據(jù)加減三倍標(biāo)準(zhǔn)偏差進(jìn)行計(jì)算的,所以在預(yù)分析階段,就要關(guān)注其結(jié)果的可能分布情況。
5.3 理化項(xiàng)目審閱
任何化學(xué)物質(zhì)都具有自己獨(dú)特的物理性質(zhì)和化學(xué)性質(zhì)。物理性質(zhì)是指不經(jīng)過化學(xué)變化就能體現(xiàn)出來的性質(zhì),如外觀、熔點(diǎn)和密度等;化學(xué)性質(zhì)是物質(zhì)經(jīng)過化學(xué)變化才能體現(xiàn)出來的性質(zhì),如閃點(diǎn)、燃點(diǎn)、可燃性和爆炸性等。物理性質(zhì)和化學(xué)性質(zhì)簡稱“理化性質(zhì)”或“物化性質(zhì)”。
和五批次GLP報(bào)告不同,各國和地區(qū)對(duì)理化性質(zhì)報(bào)告的要求差別很大,這就要求委托方在進(jìn)行理化試驗(yàn)項(xiàng)目的安排前,必須先設(shè)計(jì)好要登記的國家及地區(qū)。此外,在審閱理化項(xiàng)目中,還需要關(guān)注如下問題。
5.3.1 理化性質(zhì)的導(dǎo)則
理化性質(zhì)有很多國際通用的導(dǎo)則文件,普遍使用的有經(jīng)濟(jì)合作與發(fā)展組織(OECD)、歐盟(EU)、美國環(huán)境保護(hù)署(EPA)及國際農(nóng)藥分析協(xié)作委員會(huì)(CIPAC)方法,它們之間既有聯(lián)系也有不同。GLP實(shí)驗(yàn)室在進(jìn)行項(xiàng)目前需要仔細(xì)核對(duì)。
很多指南對(duì)某個(gè)理化性質(zhì)給出了多種測(cè)試方法,一般情況下可酌情選擇其中一種。比如密度的測(cè)量有8種方法,蒸氣壓的測(cè)定有6種方法等。在使用不同的分析方法時(shí),要注意指南對(duì)應(yīng)的檢測(cè)范圍是否合適。
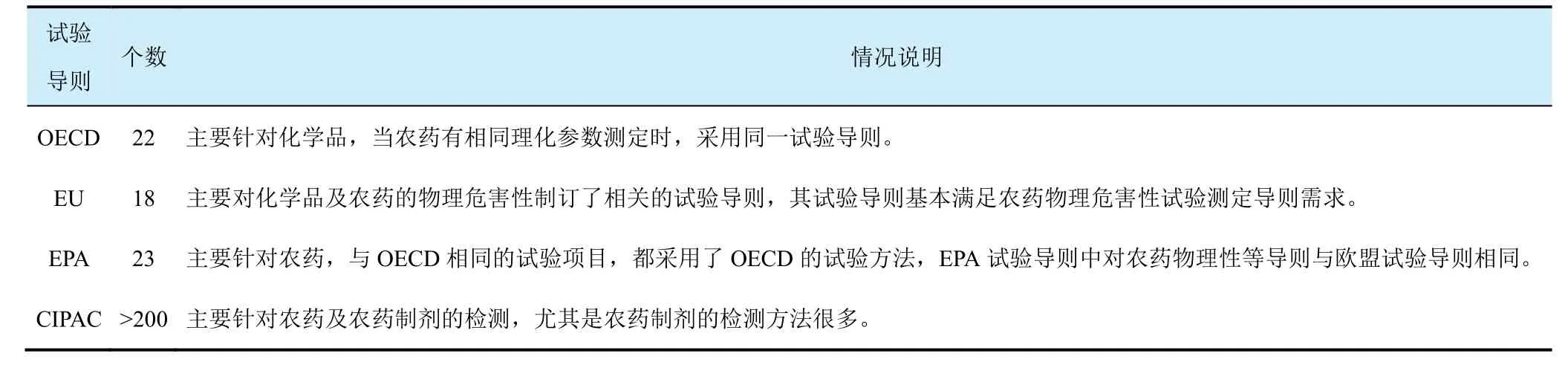
表2 理化性質(zhì)的國際指南文件
5.3.2 和文獻(xiàn)值不一致的情況
在實(shí)施理化性質(zhì)測(cè)定中,實(shí)驗(yàn)室和委托方有時(shí)會(huì)發(fā)現(xiàn),GLP報(bào)告中的數(shù)據(jù)和文獻(xiàn)報(bào)道值相差很大,這一方面可能是由于不同實(shí)驗(yàn)室使用了不同的分析方法;另外一方面,不同實(shí)驗(yàn)室之間的操作也會(huì)引起一定的差異。尤其是數(shù)值較小的檢測(cè)項(xiàng)目,如蒸氣壓,不同的分析方法測(cè)定的結(jié)果差異很大。在遇見結(jié)果差異很大的情況下,實(shí)驗(yàn)室應(yīng)該首先進(jìn)行自查,看是否屬于自身操作引起的誤差,并合理分析偏差存在的原因。
5.4 毒理項(xiàng)目審閱
毒理試驗(yàn)項(xiàng)目可以簡單分為生態(tài)毒理和哺乳動(dòng)物毒理。生態(tài)毒理試驗(yàn)是研究各種有毒有害因素,尤其是化學(xué)環(huán)境污染物對(duì)生態(tài)環(huán)境影響及其對(duì)人類之外其他生物種類包括動(dòng)物、植物和微生物等的損害作用。哺乳動(dòng)物毒理試驗(yàn)是研究外來化合物對(duì)生物體的損害作用及其機(jī)理。哺乳動(dòng)物毒理試驗(yàn)主要是應(yīng)用動(dòng)物來進(jìn)行,所觀察到的毒性反應(yīng)與人體有很多相似之處,但有些方面也有很大差異。因此,一定要進(jìn)行綜合分析,不能簡單外推于人。
不同國家和地區(qū)對(duì)毒理試驗(yàn)的要求差別很大。目前只有巴西對(duì)生態(tài)毒理項(xiàng)目有要求,墨西哥和泰國等國家要求急性毒理試驗(yàn),巴西、阿根廷和中國臺(tái)灣除了急性毒性試驗(yàn)外,還要求致突變?cè)囼?yàn)。因此,委托方必須要確認(rèn)好要登記地區(qū)的要求。此外,毒理試驗(yàn)中還應(yīng)該注意以下問題。
5.4.1 試驗(yàn)導(dǎo)則
絕大多數(shù)國家和地區(qū)都認(rèn)可按照OECD的試驗(yàn)導(dǎo)則進(jìn)行的GLP試驗(yàn),但對(duì)于同一個(gè)試驗(yàn),OECD也有多種指南文件,比如急性經(jīng)口毒性試驗(yàn)方法,就有 OECD TG420-固定劑量法(Fixed dose procedure,F(xiàn)DP)、OECD TG423-分級(jí)試驗(yàn)法(Acute toxic class method,ATC)和 OECD TG425-序貫法或上下移位法(Up-and-down procedure,UDP)。其中OECD TG423為大多數(shù)國家接受的方法。GLP實(shí)驗(yàn)室和委托方都要清楚這些要求及區(qū)別所在。
此外,由于毒理試驗(yàn)導(dǎo)則的更新較為頻繁,試驗(yàn)室需要隨時(shí)關(guān)注OECD網(wǎng)站上的更新,了解最新要求,避免使用已廢止的導(dǎo)則進(jìn)行GLP試驗(yàn)。
5.4.2 體外替代試驗(yàn)的新要求
因動(dòng)物福利及動(dòng)物試驗(yàn)方法的不確定性等原因,現(xiàn)在各國正推進(jìn)毒性測(cè)試替代方法的進(jìn)程。即使用沒有知覺的試驗(yàn)材料代替活體動(dòng)物,或使用低等動(dòng)物代替高等動(dòng)物進(jìn)行試驗(yàn)。目前歐盟和巴西已經(jīng)做出了相應(yīng)的要求,因此實(shí)驗(yàn)室和委托方應(yīng)關(guān)注各國對(duì)體外替代試驗(yàn)的要求,使報(bào)告符合登記注冊(cè)的要求。
5.4.3 毒性等級(jí)
在進(jìn)行毒理試驗(yàn)時(shí),有時(shí)候需要用統(tǒng)計(jì)軟件計(jì)算出毒性大小,通常用半致死中量(LD50)或致死中濃度(LC50)表示。毒性等級(jí)決定該農(nóng)藥產(chǎn)品是否能順利登記。當(dāng)毒性數(shù)據(jù)落在兩個(gè)等級(jí)之間的數(shù)值時(shí),要特別注意是否是統(tǒng)計(jì)方法引起的差異性。
5.4.4 文獻(xiàn)數(shù)據(jù)
一般情況下,農(nóng)藥電子手冊(cè)是境外登記人員經(jīng)常查詢的資料之一,需要注意的是農(nóng)藥電子手冊(cè)上的數(shù)據(jù)存在一定的誤差。當(dāng)審閱草稿報(bào)告時(shí),發(fā)現(xiàn)農(nóng)藥產(chǎn)品和文獻(xiàn)數(shù)據(jù)不一致的情況時(shí),要查詢?cè)摦a(chǎn)品在各國的登記材料加以佐證。
5.4.5 病理學(xué)報(bào)告
毒理病理學(xué)報(bào)告是一般毒理試驗(yàn)、嚙齒類致癌性試驗(yàn)以及生殖發(fā)育毒理試驗(yàn)等GLP試驗(yàn)需要的。毒理病理學(xué)報(bào)告要求較高,專題病理學(xué)家要具有病理學(xué)專業(yè)技能,并在毒理病理報(bào)告中簽字。毒理病理學(xué)報(bào)告中應(yīng)該有試驗(yàn)方法、數(shù)據(jù)及特殊切片或技術(shù)的詳細(xì)描述,并且有清晰的照片、圖片幫助登記機(jī)構(gòu)對(duì)結(jié)果進(jìn)行確認(rèn)。此外,報(bào)告中還需要給出給藥相關(guān)的生物學(xué)和毒理學(xué)關(guān)系等的相關(guān)科學(xué)文獻(xiàn)或試驗(yàn)背景數(shù)據(jù)等。
6 結(jié) 語
我國是農(nóng)藥生產(chǎn)和出口大國,要開拓海外市場(chǎng),優(yōu)秀的GLP報(bào)告是不可或缺的工具。GLP實(shí)驗(yàn)室和企業(yè)委托方是保證其質(zhì)量的兩大支柱。
一份優(yōu)秀的 GLP報(bào)告要有國際通用的導(dǎo)則(一般是OECD Guidelines),以及國際通用的統(tǒng)計(jì)學(xué)方法,要完全按照導(dǎo)則中要求的試驗(yàn)條件、物種、年齡、性別和環(huán)境等條件等進(jìn)行試驗(yàn)。在GLP報(bào)告中需要描述試驗(yàn)的詳細(xì)步驟、給藥方法、觀察方法、毒性反應(yīng)癥狀、體重變化、死亡時(shí)間、LD50和LD5及其可信限的計(jì)算方法和結(jié)果討論等。試驗(yàn)報(bào)告中要提供各種清晰可辨代表性譜圖或圖片,要具有科學(xué)性的令人信服的解釋。
隨著我國企業(yè)對(duì)農(nóng)藥海外登記的重視,及國內(nèi)GLP實(shí)驗(yàn)室的興起,相信中國農(nóng)藥企業(yè)能夠獲得在國際市場(chǎng)更多的話語權(quán),實(shí)現(xiàn)由農(nóng)藥大國向農(nóng)藥強(qiáng)國的轉(zhuǎn)變。
猜你喜歡
電子競(jìng)技(2020年4期)2020-07-13 09:18:06
電子競(jìng)技(2020年2期)2020-04-14 04:40:38
電子競(jìng)技(2019年22期)2019-03-07 05:17:26
電子競(jìng)技(2019年21期)2019-02-24 06:55:52
電子競(jìng)技(2019年20期)2019-02-24 06:55:35
電子競(jìng)技(2019年19期)2019-01-16 05:36:09
南方人物周刊(2017年32期)2017-10-28 22:48:36
南風(fēng)窗(2016年26期)2016-12-24 21:48:09
南風(fēng)窗(2015年22期)2015-09-10 07:22:44
南風(fēng)窗(2015年14期)2015-09-10 07:22:44
