臍帶間充質干細胞制劑的質量管理及有效性和安全性
2020-04-27 06:25:32王佃亮
轉化醫學雜志 2020年2期
王佃亮
國內外利用臍帶間充質干細胞制劑進行的動物模型試驗、臨床研究和臨床移植治療表明,不同的臍帶間充質干細胞制劑療效差別很大[1-5]。究其原因,可能與疾病種類、治療時機、使用劑量、移植途徑、個體差異等因素有關,但主要還是由干細胞制劑的質量差異導致的。造成臍帶間充質干細胞制劑質量差異的因素很多,主要包括細胞來源的組織材料、分離制備方法及過程、檢測鑒定方法及過程、傳代方法及過程、擴增方法及過程、制劑配制添加成分及配制方法和過程、冷藏或冷凍保存方法及過程、復蘇方法及過程、操作人員技能差異等。干細胞制劑的質量與干細胞治療的有效性和安全性有關,是干細胞臨床應用的關鍵。
1 臍帶間充質干細胞制劑的質量管理
臨床級的干細胞制劑以人體疾病治療為目的,其生產和管理應遵循《藥品生產質量管理規范》(good manufacturing practice,GMP)、《人體細胞治療研究和制劑質量控制技術指導原則》(國家食品藥品監督管理局2003年3月頒布)、《干細胞制劑質量控制及臨床前研究指導原則(試行)》(國家衛生計生委、食品藥品監督管理總局2015年7月頒布)、《干細胞制劑制備質量管理自律規范》(中國醫藥生物技術協會2016年10月發布)、《干細胞通用要求》(中國細胞生物學學會干細胞生物學分會2017年11月發布)等國家法規和行業標準[6-7]。在臍帶間充質干細胞制劑生產過程中,需要嚴格遵守這些法規和標準,提高質量。
1.1 制劑制備過程及所用材料的質量管理 預先制定新生兒臍帶采集及臍帶間充質干細胞分離純化、原代培養、傳代擴增、細胞鑒定、細胞系建立、冷凍保存、干細胞注射液配制和保存的標準操作及質量管理程序,建立質量標準(表1),并在符合GMP要求基礎上嚴格執行。
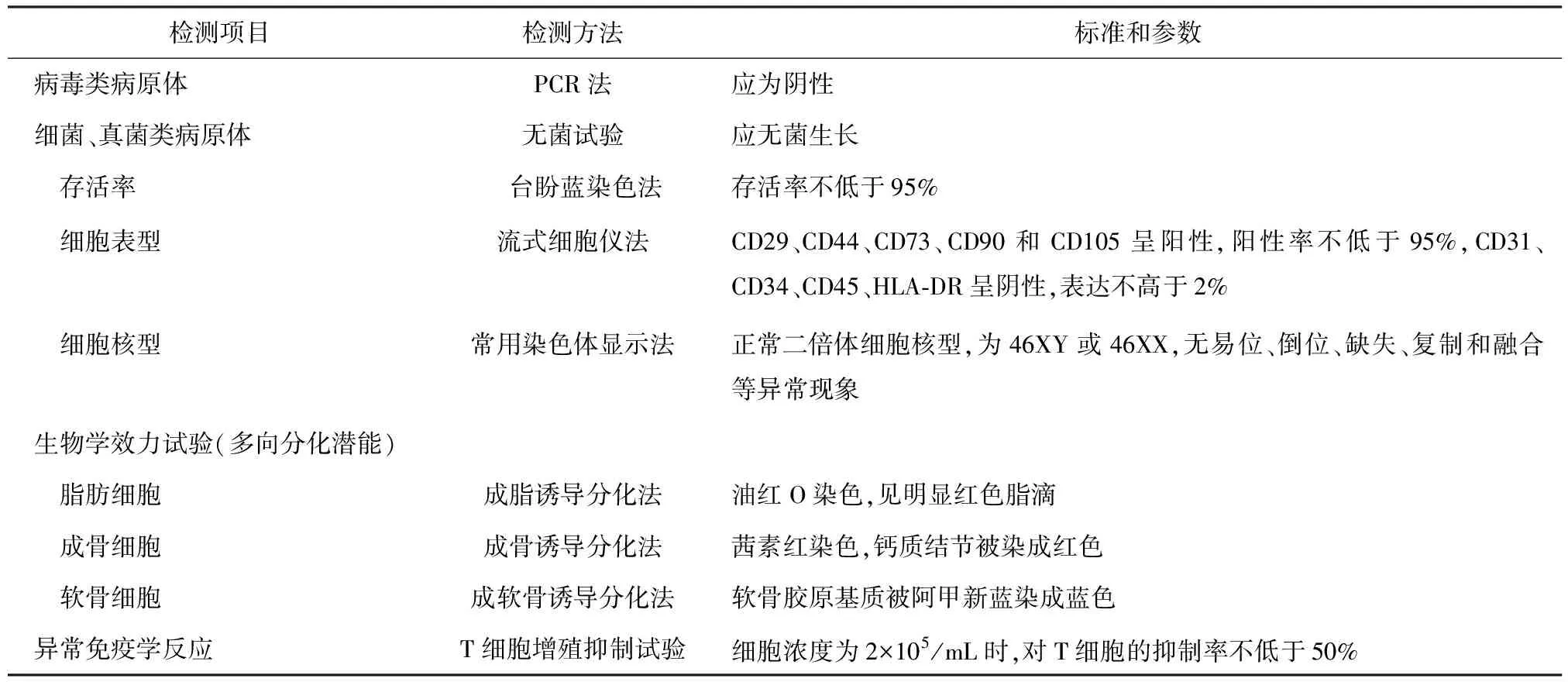
表1 臍帶間充質干細胞的質量標準
新生兒臍帶樣本的采集場所應達到Ⅱ級一般潔凈手術室要求,保證無菌采集環境。采集前,選擇健康足月產婦,保證:①獲得產婦本人、法定代表人或監護人同意,簽署知情同意書;②經檢驗產婦乙型肝炎病毒(hepatitis B virus,HBV)、丙型肝炎病毒(hepatitis C virus,HCV)、艾滋病病毒(human immunodeficiency virus,HIV)、EB病毒(Epstein-Barr virus,EBV)、巨細胞病毒(cytomegalovirus,CMV)、人類嗜T淋巴細胞病毒(human T cell lymphotropic virus,HTLV)、梅毒螺旋體、支原體、霉菌等病原體均為陰性;③產婦本人及家族成員無遺傳病、傳染病、精神病或其他重大疾病現病史和既往病史。采集時,待胎兒娩出,立刻用蘸有75%酒精的紗布擦拭臍帶,截取至少15 cm無針孔的臍帶放入保存液中,置于無菌采集袋里,標識清楚,臍帶兩端用手術線結扎。
在無菌室操作臺上,從新生兒臍帶組織剝取華通氏膠,以組織塊培養法或酶消化法分離新生兒間充質干細胞,貼壁法做原代培養。培養液中若加有牛血清、豬胰酶,除要求供應商提供企業合法生產資質、產品合格證及說明書等信息外,還要檢測牛、豬來源的危險致病病毒。傳代培養要有明確的細胞鑒別特征,細胞純度和活性達到臨床要求,并且無內外源微生物污染。經傳代和擴增培養后達到一定數量的細胞系,可以進行冷凍保存。凍存細胞需加入合適的冷凍保護液,在程序降溫條件下進行冷凍。凍存好的細胞移至液氮罐中(-196~-135 ℃)長期儲存。建立主細胞庫和工作細胞庫,減少不同批次臍帶間充質干細胞在操作過程中的變異性[8]。
干細胞培養液添加物、干細胞注射液配制所需的溶媒和添加物都要有合法的來源,生產企業有相應資質,能夠提供產品合格證和說明書等信息。干細胞制備及制劑配制的相關重要信息(如:干細胞供者的年齡、民族、血型、現病史、既往病史等;干細胞制劑的生產日期、批號、有效期、入庫時間、出庫時間等)應錄入計算機系統進行管理,核心數據信息要進行備份,以防感染計算機病毒或遭黑客襲擊丟失信息。
1.2 制劑的質量檢測 臨床移植治療用的臍帶間充質干細胞制劑,需要經過細胞檢測、制劑檢測、放行檢測以及質量復核等程序。
1.2.1 細胞檢測 包括但不限于:①細胞鑒別:通過細胞形態、遺傳學、代謝酶亞型譜分析、表面標志物、特定基因表達產物等檢測,對不同供體的臍帶間充質干細胞進行綜合的細胞鑒別。在倒置顯微鏡下觀察,體外微組織塊培養法培養的原代人臍帶間充質干細胞在第6~8天開始有少量細胞從組織塊中爬出,沿組織周圍向外呈放射狀延伸生長,吹打去除組織塊后呈旋渦狀生長,細胞呈梭形,與皮膚成纖維細胞類似,形態相對均一。原代人臍帶間充質干細胞長滿瓶底并達到90%融合時間為18~20 d。傳代培養的臍帶間充質干細胞生長速度比原代快,24 h內可見少量貼壁的圓型細胞,逐漸呈克隆式生長,到3 d左右時,大多數呈梭形,部分呈多邊或多角形,長滿瓶底后基本呈梭形,旋渦狀生長。根據其生長形態和特征,結合組織來源,可初步鑒別臍帶間充質干細胞。在通過表面標志物等分析,可明確鑒定。正常二倍體細胞核型,為46 XY或46 XX;CD29、CD44、CD73、CD105、CD90陽性,陽性率大于95%;HLA-DR、CD34、 CD31、CD45陰性,表達小于2%等。②存活率和生長活性:采用多種細胞生物學活性檢測方法,如活細胞計數、細胞倍增時間、細胞周期、克隆形成率、端粒酶活性等判斷細胞活性和生長情況。存活率不低于95%;體外擴增人臍帶間充質干細胞潛伏期為1~2 d,對數生長期為3~7 d,第8天以后長滿瓶底轉入平臺期,生長停滯,傳代培養增殖倍數為5~6倍;人臍帶間充質干細胞85%以上處于G0/G1期,G2/M期占5%以下,S期細胞占10%以下。③純度、均一性及最低裝量:通過肉眼觀察、檢測細胞表面標志物和遺傳多態性及特定生物學活性等,對制劑進行細胞純度或均一性的檢測;用最低裝量檢查法檢查最低裝量。標準和參數為白色均勻懸濁液,無明顯沉淀,無異物。每劑的最低裝量應為(20±2)mL。④無菌試驗和細菌、真菌、支原體、梅毒螺旋體檢測:依據2015版《中華人民共和國藥典》中生物制品無菌試驗和支原體檢測規程,對細菌、真菌、支原體污染進行檢測。標準和參數為:應無菌生長;細菌、支原體、真菌、梅毒螺旋體檢測為陰性。⑤細胞內外源致病因子檢測:應結合體內和體外方法,根據每一細胞制劑的特性進行人源及動物源性特定致病因子,利用PCR技術、酶聯免疫法等進行檢測。如使用過牛血清,須進行牛源特定病毒的檢測;如使用胰酶等豬源材料,應至少檢測豬源細小病毒。另外,還應檢測逆轉錄病毒(如HIV)。標準和參數為:HBV、HCV、HIV、EBV、CMV、HTLV、牛海綿狀腦病病毒、豬細小病毒等檢測應為陰性。⑥內毒素檢測:依據2015版《中華人民共和國藥典》中內毒素檢測規程中的凝膠法,對內毒素進行檢測。內毒素檢測標準為≤50 EU。⑦異常免疫學反應檢測:檢測異體來源干細胞對人總淋巴細胞增殖和對不同淋巴細胞亞群增殖能力的影響,或對相關細胞因子分泌的影響,分析可能引起的異常免疫反應。判定標準為間充質干細胞濃度為2×105/mL時,對T淋巴細胞增殖抑制率不低于50%。⑧致瘤性和促瘤性檢驗:對于異體來源的干細胞制劑或經體外復雜操作的自體干細胞制劑,須通過免疫缺陷動物體內致瘤試驗,檢驗細胞的致瘤性。判定標準為無致瘤性。⑨生物學效力試驗:通過檢測干細胞分化潛能、誘導分化細胞的結構和生理功能、對免疫細胞的調節能力、分泌特定細胞因子、表達特定基因和蛋白等功能,判斷干細胞制劑與治療相關的生物學有效性。對間充質干細胞,無論何種來源,應進行體外多種類型細胞(如成脂肪細胞、成軟骨細胞、成骨細胞等)分化能力的檢測,以判斷其細胞分化的多能性。除此以外,作為特定生物學效應試驗,應進行與其治療適應證相關的生物學效應檢驗。判定標準為應具有相應生物學功能。⑩細胞培養基及其他添加成分殘余量的檢測:對臍帶間充質干細胞制備過程中殘余的、影響臍帶間充質干細胞制劑質量和安全性的成分,如牛血清白蛋白、抗生素、細胞因子等進行檢測。注射劑中牛血清殘留量檢測使用牛血清白蛋白酶聯免疫法或間接凝集法,判定標準為牛血清殘留量不得大于50 ng/mL;細胞注射劑抗生素殘留量檢測采取培養法,標準為陰性;人表皮生長因子采用ELISA的方法檢測,應不大于100 pg/mL。
1.2.2 制劑檢測 臍帶間充質干細胞制劑(注射液)是臍帶間充質干細胞與0.9%的生理鹽水或復方電解質注射液(基礎溶媒)、L-谷氨酰胺或腺苷(能源物質)、N-乙酰半胱氨酸或亞硒酸鈉(抗氧化劑)、胰島素(活性生長因子)、肝素鈣(抗凝劑)、甘油或二甲亞砜(滲透性細胞內冷凍保護劑)、人血白蛋白或葡聚糖(非滲透性細胞外冷凍保護劑)等成分配制而成。它的檢測是在干細胞檢測的基礎上,進一步檢測制劑中臍帶間充質干細胞的形態、存活率、生長活性等,檢測制劑的純度和均一性、微生物病原體、內毒素、異常免疫反應、致瘤性、促瘤性、生物學效力等。臍帶間充質干細胞注射劑的包裝必須采取符合2015版《中華人民共和國藥典》的包裝要求,包裝材料必須無菌、無色透明、不吸附細胞,不影響細胞活性;制備的細胞注射劑成品外觀肉眼觀察應為白色均勻混懸液,無明顯的沉淀物、無異物;每劑的最低裝量應為(20±2)mL。
1.2.3 放行檢測 針對臍帶間充質干細胞制劑的特性,制定放行檢測項目和標準。放行檢測項目應能在相對短的時間內,反映臍帶間充質干細胞制劑的質量和安全信息。
1.2.4 質量復核 由專業細胞檢驗機構或實驗室進行臍帶間充質干細胞制劑的質量復核檢驗,并出具正規的檢驗報告。
1.3 制劑的穩定性 臍帶間充質干細胞制劑的穩定性是指制劑在儲存(液氮凍存和細胞植入前的臨時存放)和運輸過程中的物理、化學和生物學性質的改變。檢測項目包括細胞形態、活性、存活率、密度、純度、無菌性等。穩定性檢測的關鍵是制劑中臍帶間充質干細胞的數量和活性的改變,直接影響到臍帶間充質干細胞治療的效果。
根據臍帶間充質干細胞制劑穩定性檢測結果,確定制劑的保存液成份與配方、保存和運輸條件、有效期,同時確定與有效期相適應的運輸容器和工具,以及合格的細胞凍存設施和條件。
2 臍帶間充質干細胞制劑的有效性
臍帶間充質干細胞制劑的有效性可利用細胞模型和動物模型在臨床前研究階段進行評估[9]。①細胞模型通過檢測臍帶間充質干細胞的分化潛能、誘導分化細胞的結構和生理功能、對免疫細胞的調節能力、分泌特定細胞因子、表達特定基因和蛋白等功能,判斷臍帶間充質干細胞制劑與治療相關的生物學有效性。對臍帶間充質干細胞進行體外多種類型細胞(成脂肪細胞、成軟骨細胞、成骨細胞等)分化能力的檢測,以判斷細胞分化的多能性。作為特定生物學效應試驗,應進行與治療適應證相關的生物學效應檢驗。隨著研究的進展,針對臨床治療的適應證,會不斷研究更新生物學效應檢測方法。如研究介導臨床治療效應的關鍵基因或蛋白的表達,并以此為基礎提出與預期的生物學效應相關的替代性生物標志物。②動物模型用于觀察植入的臍帶間充質干細胞或其分化產物改變模型中疾病的病理進程;研究臍帶間充質干細胞的歸巢能力和免疫調節功能;通過分析臍帶間充質干細胞植入后特定細胞因子和(或)特定基因表達情況,提出替代性生物學效應標志物。
國內外眾多臨床研究和治療病例表明,臍帶間充質干細胞可用于自身免疫性疾病、退行性疾病、神經性疾病、衰老性疾病、遺傳缺陷、組織器官損傷、放射病、炎癥等病癥的治療[10-12],包括兒童移植物抗宿主病、克羅恩病、創傷、缺血性損傷、造血功能障礙、骨及軟骨損傷、神經損傷、肌營養不良、糖尿病及其并發癥、骨關節炎、類風濕性關節炎、系統性紅斑狼瘡、肝硬化、肝纖維化、酒精性肝病、肝功能衰竭、腎功能衰竭、急性心肌梗死、腦梗死、腫瘤、視網膜黃斑變性、帕金森病、老年癡呆癥、肌萎縮側索硬化癥、系統性硬化癥、原發性干燥綜合征、皮肌炎等難治性疾病。此外,臍帶間充質干細胞是比較理想的組織工程種子細胞,可用于體外或體內再生組織器官,進行臨床治療。
3 臍帶間充質干細胞制劑的安全性
臍帶間充質干細胞制劑的安全性包括生物學安全、病原生物安全、倫理學安全等[9]。臍帶間充質干細胞來源于產婦產后的臍帶組織,通常情況下是作為醫療廢物丟棄的,從臍帶組織中分離提取干細胞不存在倫理爭議,可以用于臨床治療,在倫理學上是安全的,這一點與某些胚胎干細胞不同[13-15]。
臍帶間充質干細胞制劑的生物學安全主要是指毒性反應、異常免疫學反應、非預期分化、致瘤性和促瘤性等。①毒性反應:通過合適的動物實驗模型,觀察臍帶間充質干細胞制劑各種可能的毒性反應,譬如,細胞植入時和植入后的局部和整體的毒性反應。②異常免疫學反應:通過體外和動物試驗評價其異常免疫反應,包括對不同免疫細胞亞型及相關細胞因子的影響。③非預期分化:包括非靶細胞分化或非靶部位分化,可利用特定的檢測技術,在體內動物試驗中研究、評估和監控臍帶間充質干細胞非預期分化的可能性。④致瘤性和促瘤性:目前,普遍認為間充質干細胞“不致瘤”或具有“弱致瘤性”,但不排除對已存在腫瘤的“促瘤性”作用,應設計相應的試驗方法,以判斷制劑的“促瘤性”。對高代次或經過體外復雜處理和修飾的臍帶間充質干細胞制劑,應當進行動物致瘤性和促瘤性評估。可選擇合適的動物實驗模型,使用合適數量的干細胞、合理的植入途徑和足夠長的觀察期,以有效評價制劑的致瘤性和促瘤性。
臍帶間充質干細胞制劑的病原生物安全是指攜帶和傳播細菌、病毒、支原體、衣原體、真菌、寄生蟲的風險和內毒素殘留等。一些常見的病原體,如HBV、HCV、HIV、EBV、CMV、HTLV、梅毒螺旋體、支原體、霉菌、內毒素等,已經建立了實驗室檢測方法和相應的質量標準,基本可以滿足臨床要求。隨著新的病原體被發現,將來會增加新的檢測項目,確保臍帶間充質干細胞制劑不含任何內部的和外源的病原體。
通過制定符合臨床治療要求的臍帶間充質干細胞制劑生產、儲存、運輸的質量標準,建立標準化的質量檢測方法和流程,進行嚴格的質量管理,并不斷完善這些質量標準、質量檢測方法和質量管理流程,臍帶間充質干細胞制劑可以達到臨床治療要求。
迄今,國內外大量臨床研究和治療病例表明,臍帶間充質干細胞治療是安全的,除極少數患者輕度腰部酸痛、發熱、頭昏、頭痛外,無其他任何嚴重不良反應。但是,目前仍有一些患者不宜進行臍帶間充質干細胞治療。這些患者是:晚期惡性腫瘤尤其是腦腫瘤患者;休克或全身衰竭生命體征不正常及不配合檢查者;合并心、肺、肝、腎等重要臟器功能障礙者;全身感染或局部嚴重感染抗感染康復前;凝血功能障礙者;血清學檢查(如艾滋病、乙肝、梅毒等)陽性;高度過敏體質或有嚴重過敏史者;非神經系統疾患或尚未明確診斷者;其他不適于臍帶間充質干細胞移植的人群。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
現代畜牧科技(2021年4期)2021-07-21 06:13:00
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
福建基礎教育研究(2019年1期)2019-09-10 07:22:44
福建基礎教育研究(2019年1期)2019-05-28 08:39:49
數學物理學報(2017年2期)2017-06-05 09:12:30
海峽科技與產業(2016年3期)2016-05-17 04:32:12
