新型吸附劑-固相萃取-超高效液相色譜-串聯質譜法測定魚塘水中21種常見農藥
2020-04-24 10:43:38徐樞雅
理化檢驗-化學分冊 2020年3期
劉 民,徐樞雅
(揚州市公安局刑警察支隊,揚州225009)
魚塘投毒造成大量魚蝦死亡的案例每年都有發生,由于魚塘水體積較大,水中毒藥含量極低,要想取得較可靠的結果,必須對大量魚塘水進行前處理。傳統液液提取法或固相萃取小柱法不僅操作煩瑣、耗時,還得不到滿意結果,在此類案件中常束手無策。
本工作以辛烷基硅烷鍵合硅膠(C8)、十八烷基硅烷鍵合硅膠(C18)、聚二乙烯苯多孔小球GDX403[1]、大孔吸附樹脂 X-5 等4 種填料混合而成的固相萃取吸附劑對常見的21 種農藥進行富集[2-4],然后采用超高效液相色譜-串聯質譜法(UPLC-MS/MS)進行測定。本工作采用的前處理方法(新型吸附劑-固相萃取)簡潔、方便,具有一定創新性,可為實驗室對大體積水樣的前處理提供技術指南。
1 試驗部分
1.1 儀器與試劑
AB Qtrap 4000型質譜儀、島津LC-30AD 型超高效液相色譜儀;Turbo VapⅡ型自動氮吹濃縮儀;HY-2 型振蕩器;Vortex-Genie 2 型漩渦混合器;MILLIPAK 型 純 水 儀 ;Maximum Vacuum 型 真空泵。
21種農藥標準儲備溶液:分別吸取21種農藥標準品各適量,或用甲醇稀釋,配制成1.0 g·L-121種農藥標準儲備溶液。
21種農藥混合標準溶液:取上述21種農藥標準儲備溶液各100μL 至10 m L 容量瓶中,用甲醇定容,配制成10 mg·L-1混合標準溶液。試驗中所用其余質量濃度的混合標準溶液均從上述混合標準溶液用甲醇稀釋而得。
內標物標準儲備溶液:稱取普羅迪芬鹽酸鹽(SKF525A)和苯巴比妥適量,用甲醇配制成兩種質量濃度均為1.0 g·L-1的內標物標準儲備溶液待用。
內標物混合標準溶液:分別取50μL SKF525A標準儲備溶液和200μL 苯巴比妥標準儲備溶液至100 m L容量瓶中,用甲醇定溶,配制成含SKF525A 0.5 mg·L-1和苯巴比妥2 mg·L-1的內標物混合標準溶液。
硫丹Ⅱ、氯硝柳胺、硫丹硫酸酯、魚藤酮、甲氰菊酯、溴氰菊酯、氰戊菊酯、氯氰菊酯等21種標準品均為1.0 g·L-1或2.0 g·L-1的標準品溶液;內標物SKF525A 的純度為95%,內標物苯巴比妥的純度為98%。
乙腈、甲醇、甲酸均為色譜純;乙酸銨為分析純;C8(58~75μm)、C18(58~75μm)、聚二乙烯苯多孔小球GDX-403(150~180μm)、大孔吸附樹脂X-5(1.08~2.00 mm,含水量65%~75%);試驗用水為超純水。
1.2 儀器工作條件
1.2.1 色譜條件
InertSustain C18色譜柱(2.1 mm×50 mm,2μm);柱溫35℃;流量400μL·min-1;進樣量5μL。
1)洗脫條件1 流動相:A 為含0.1%(體積分數,下同)甲酸的20 mmol·L-1乙酸銨溶液,B為乙腈。梯度洗脫程序:初始流動相B 為10%,保持1 min;2~10 min 時,B 由10%升至70%;10~11 min時,B 由70%升至90%,保持2 min;13~14 min時,B由90%降至10%,保持2 min。
2)洗脫條件2 流動相:A 為含0.1%甲酸的20 mmol·L-1乙酸銨溶液,B為甲醇。梯度洗脫程序:初始流動相B 為90%,保持1 min;1~2 min時,B由90%升至96%,保持2 min;4~5 min時,B由96%降至90%,保持2 min。
3)洗脫條件3 流動相:A 為20 mmol·L-1乙酸銨溶液,B為甲醇。梯度洗脫程序:初始流動相B為50%,保持2 min;2~3 min時,B由50%升至95%,保持2 min;5~6 min 時,B 由 95%降至50%,保持1 min。
1.2.2 質譜條件
電噴霧離子源(ESI),正離子和負離子模式,多反應監測(MRM)模式,霧化氣和脫溶劑氣為氮氣,碰撞氣為氮氣,氣簾氣壓力172.5 k Pa;離子噴射電壓5 500 V(-4 500 V);霧化氣溫度500℃;霧化氣壓力345.0 k Pa,輔助加熱氣壓力345.0 k Pa;入口電壓10 V(-10 V),碰撞氣出口電壓15 V(-15 V)。對其余質譜參數進行優化[5],見表1。
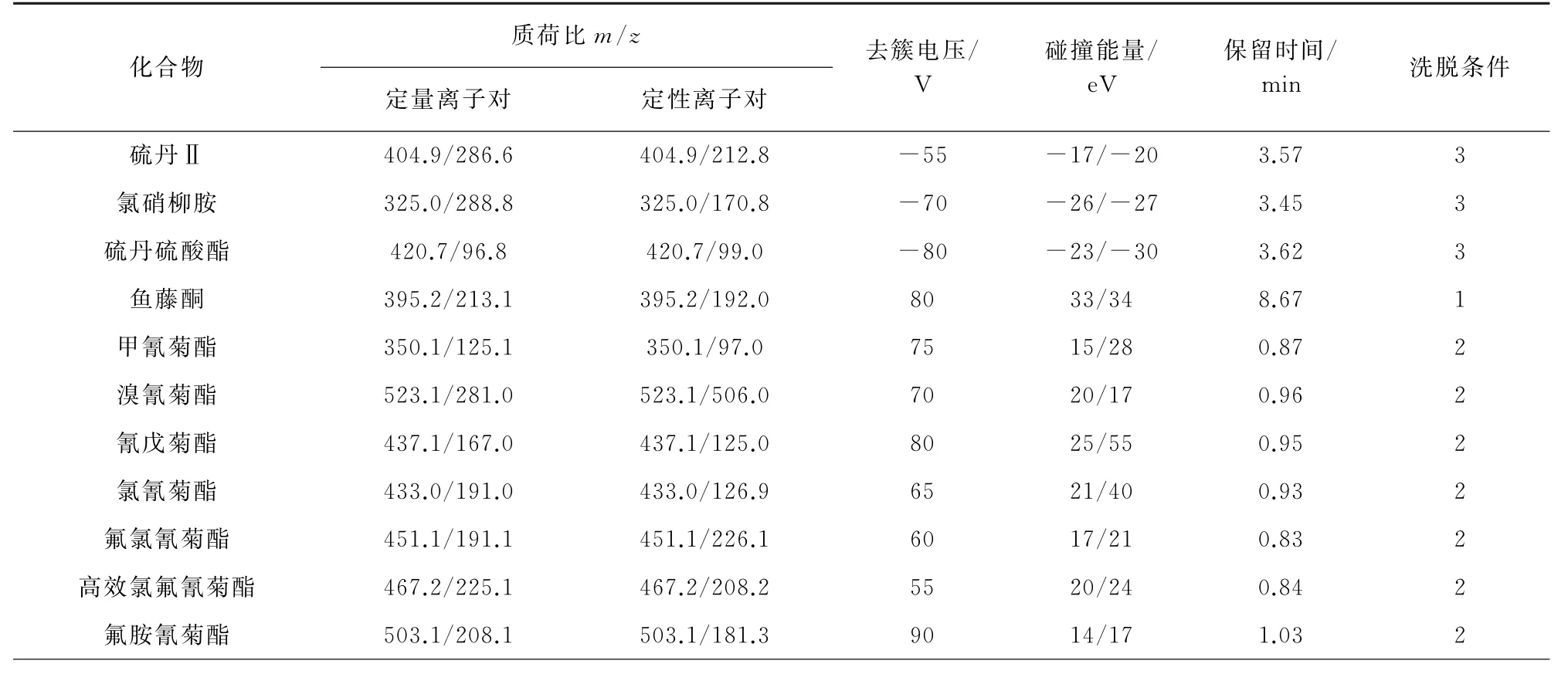
表1 21種常見農藥及2種內標物的質譜參數Tab.1 MS parameters of the 21 common pesticides and the 2 internal standards
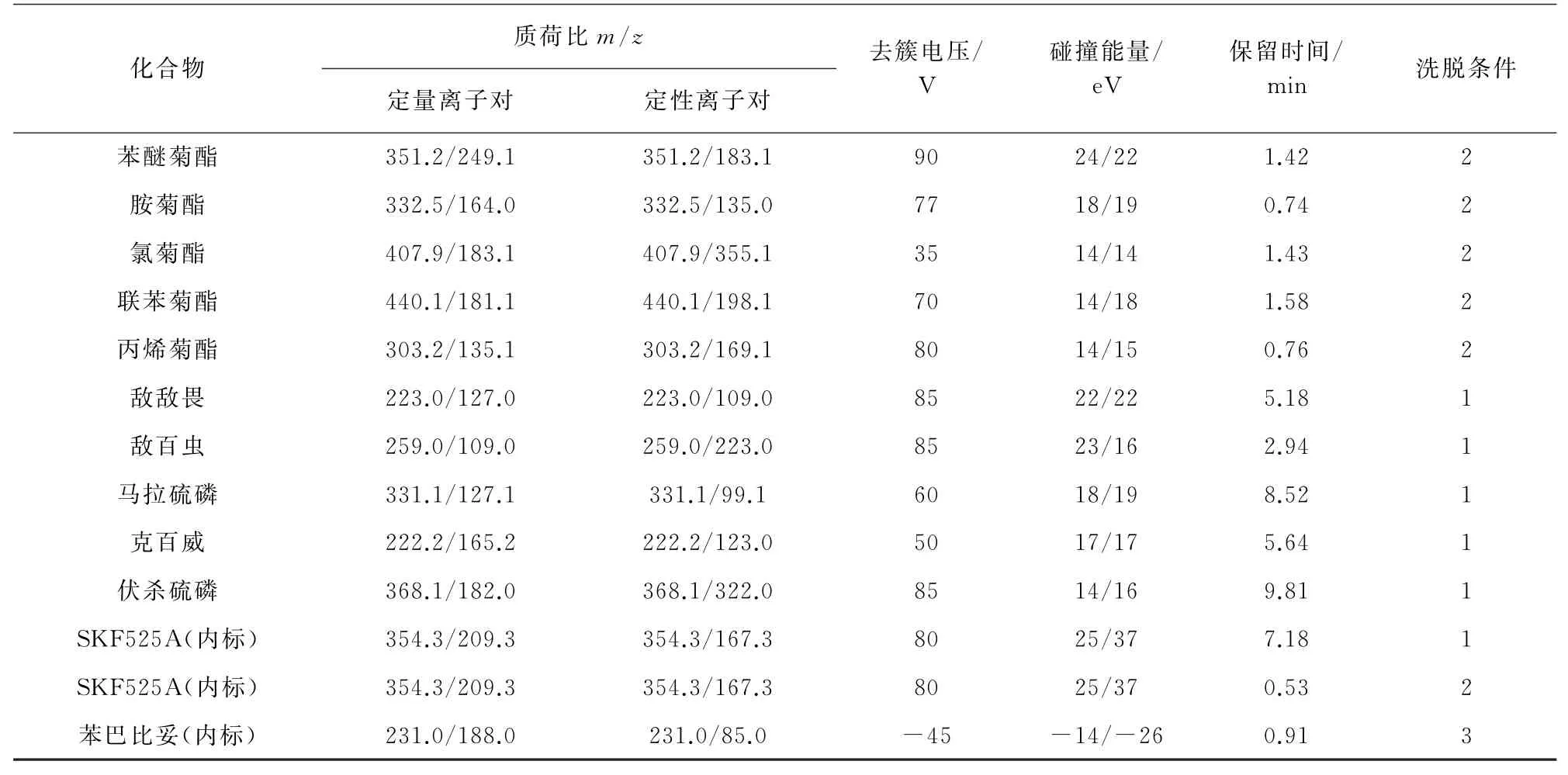
表1(續)
1.3 試驗方法
取魚塘水樣4 L于5 000 m L燒瓶內,添加內標物混合標準溶液100μL;稱取由C8、C18、GDX403和X-5按1∶2∶1∶8的質量比混合而成的吸附劑6 g,經10 m L 甲醇浸泡活化、過濾后,加入到上述水樣中;將燒瓶固定在振蕩器上振蕩約1 h,用布氏漏斗抽濾,將收集到的固相吸附劑連同濾紙一起放入250 m L錐形瓶中,加入5 g無水硫酸鎂和苯、乙酸乙酯各100 m L,振蕩10 min后,用1.0μm 微孔有機濾膜過濾,將濾液吹氮至近干,加入1 m L甲醇定容、0.22μm 微孔有機濾膜過濾,濾液按儀器工作條件進行測定[6]。
2 結果與討論
2.1 色譜條件的選擇
試驗發現,流動相梯度變化大,以乙腈作流動相B,按洗脫條件1進行洗脫時,可分離的農藥種類較多,適用魚藤酮等6種目標物的測定;但按洗脫條件1進行洗脫時,甲氰菊酯等12種擬除蟲菊酯類農藥分離效果差甚至不出峰;而使用色譜條件2測定時,擬除蟲菊酯類農藥靈敏度高、峰形好,說明含0.1%甲酸的20 mmol·L-1乙酸銨溶液-甲醇的出峰效果優于含0.1%甲酸的20 mmol·L-1乙酸銨溶液-乙腈流動相[7],試驗還發現,梯度洗脫時,初始流動相中甲醇含量要高[8],否則擬除蟲菊酯類農藥不出峰。洗脫條件3的流動相B為50%~95%,有效地避免了目標物出峰時間過早或過遲,適用于硫丹Ⅱ等3種農藥的測定。因此,試驗選擇了3種洗脫條件,色譜圖見圖1。
2.2 質譜條件的選擇
硫丹為實際魚塘投毒案件中最常見毒物之一,前期大量UPLC-MS/MS試驗結果表明[9],硫丹Ⅰ和硫丹Ⅱ的定量離子對和定性離子對、去簇電壓(DP)、碰撞能量(CE)和保留時間均相同,雖然同含量硫丹Ⅰ和硫丹Ⅱ的相同定量離子對或相同定性離子對的峰強度有差異,但硫丹Ⅰ的定性離子對/定量離子對和硫丹Ⅱ的定性離子對/定量離子對的豐度比一致,硫丹Ⅱ的定性離子對或定量離子對峰強度更高。綜合考慮,試驗選擇硫丹Ⅱ作為分析對象。
21種常見農藥中,硫丹Ⅱ等3種農藥因在電離時更容易失去質子,需采用負離子模式進行測定。內標物苯巴比妥也選擇負離子模式。
因擬除蟲菊酯類農藥測定難度較大,本試驗采用針泵注射方式,對12種擬除蟲菊酯類農藥在正離子和負離子模式下進行一級全掃描,發現溴氰菊酯等7種擬除蟲菊酯類農藥在正離子模式下的分子離子峰豐度最大。因此,這7種物質的母離子選擇了],剩下5種擬除蟲菊酯類農藥和其他6種農藥及內標物SKF525A 的母離子均選擇了[M+H+]。
2.3 固相萃取劑的選擇
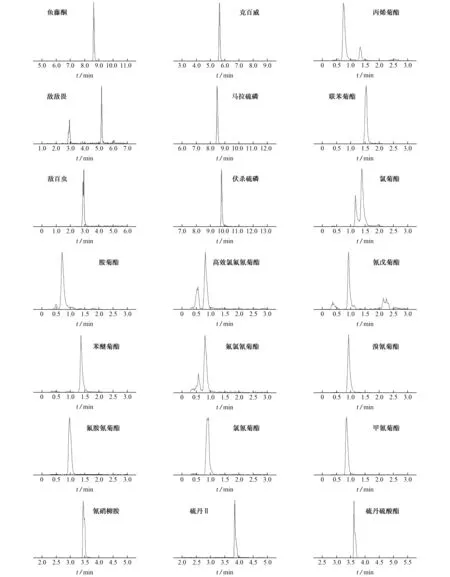
圖1 21種常見農藥的色譜圖Fig.1 Chromatograms of the 21 common pesticides
本工作采用由4種常見填料相混的混合物進行試驗[10-12],C8、C18、GDX403 和 X-5 按1∶2∶1∶8的質量比混合。分別稱取C8、C18、GDX403各2 g,X-5、混合填料各6 g,考察了 C8、C18、GDX403、X-5和混合填料等5種填料對19種目標物的提取效果,具體方法為:取空白水樣4 L 共10份,各添加混合標準溶液200μL,得到水樣中19種目標物的質量濃度均為0.5μg·L-1,再加入內標物混合標準溶液100μL;然后分別加入5種填料,每種填料有兩個平行樣,按試驗方法進行測定,19種目標物定量離子對峰面積為A1。另取空白水樣4 L 共10份,不添加混合標準樣品,分別加5種填料,每種填料有兩個平行樣,在濾液吹氮縮至近干后,加1 m L含100μL混合標準溶液及100μL 內標物混合標準溶液的甲醇溶液定容,在儀器工作條件下測定,19種目標物定量離子對峰面積為A2。每次操作前,均取相應固相填料加入空白水樣(4 L)進行空白比對,確保無干擾、無殘留。回收率為A1/A2,如果2個平行樣回收率的相對差值在5%之內,說明測定結果有效,取2次結果的平均值;否則,應重新測定。結果見表2。
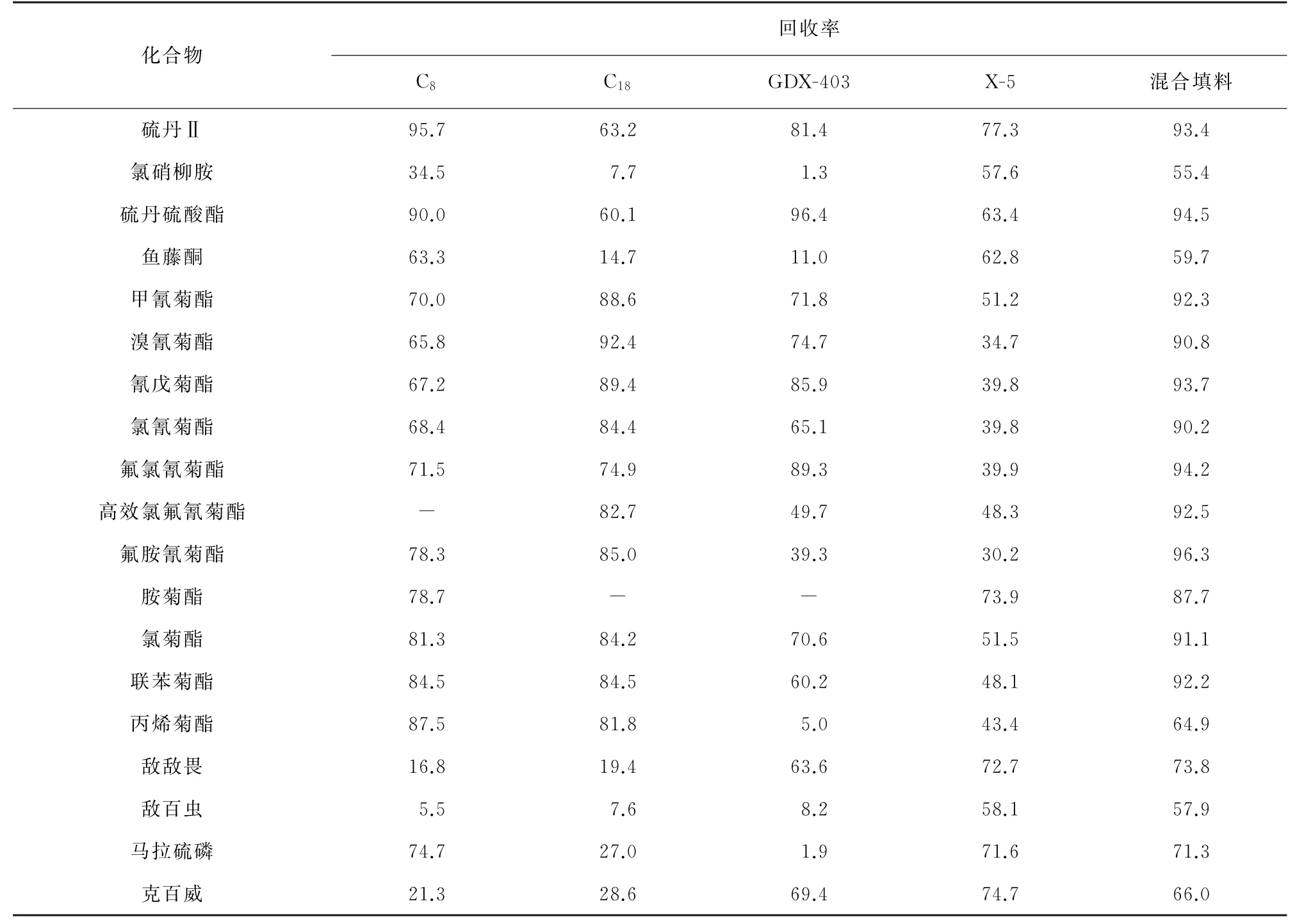
表2 使用5種不同固相填料作為吸附劑時19種常見農藥的回收率Tab.2 Recovery of 21 common pesticides with 5 different fillers as adsorbents %
由表2 可知:當使用單一填料進行提取時,19種常見農藥的回收率有高有低;當使用混合填料進行提取時,19種常見農藥的回收率與其他4種單一填料中回收率最高的相接近。因魚塘投毒案件中毒物種類的不確定性,在檢驗中使用單一填料進行前處理,往往出現一種或多種目標物回收率極低甚至無法檢出等情況,不能滿足實際要求。
2.4 工作曲線
取空白水樣4 L,添加混合標準溶液,配制成質量濃 度 為 0.5,0.25,0.1,0.05,0.025,0.01,0.005μg·L-1混合標準溶液系列,按試驗方法進行測定。
21種目標物定量離子對峰面積為A3、內標定量離子對峰面積為A4,每次試驗均用混合填料對空白水樣進行空白比對,確保無干擾、無殘留。以21種常見農藥的質量濃度為橫坐標,其對應的A3/A4結果為縱坐標繪制工作曲線,線性范圍、線性回歸方程及相關系數見表3。
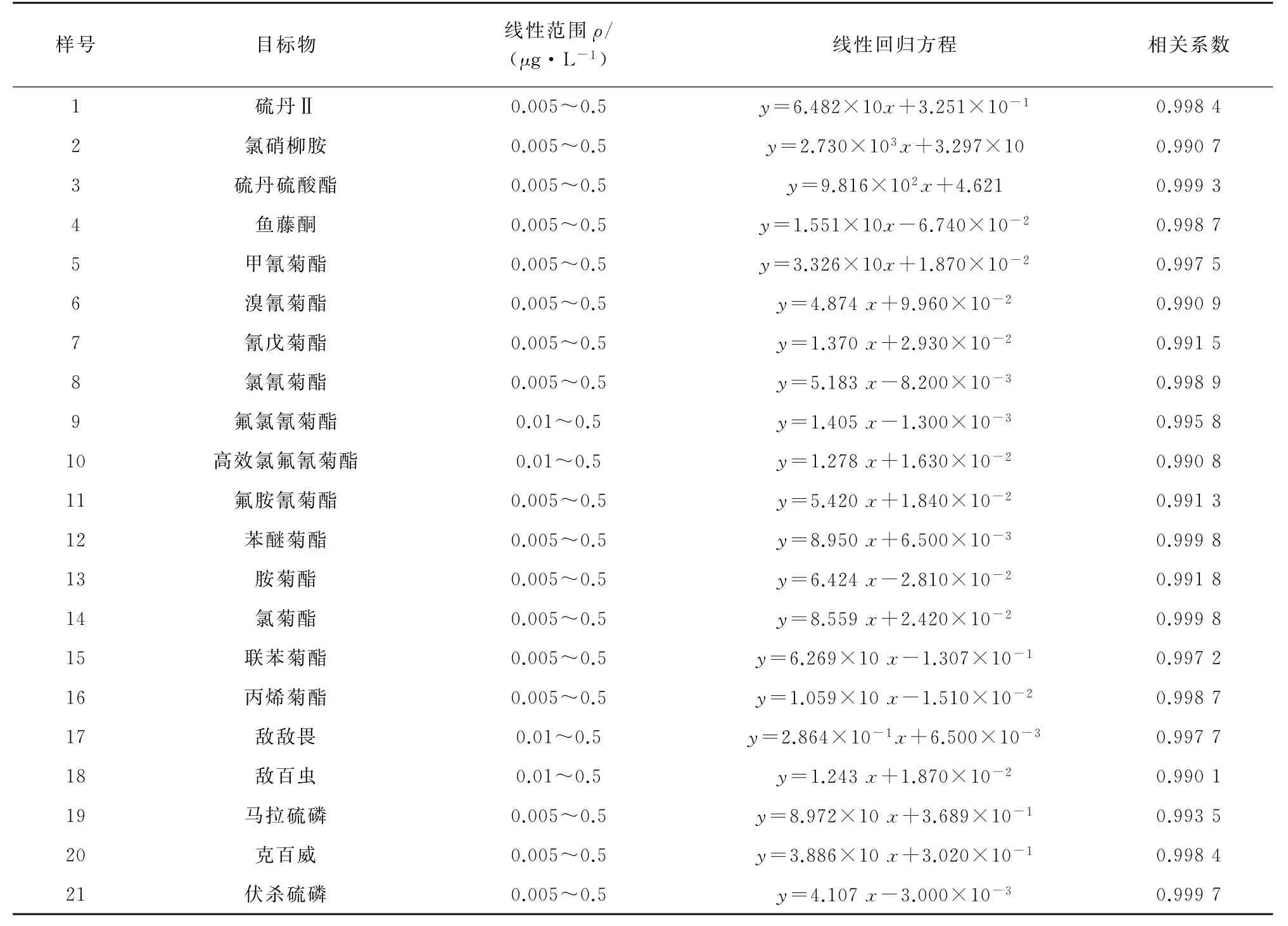
表3 線性范圍、線性回歸方程和相關系數Tab.3 Linear ranges,linear regression equations and correlation coefficients
2.5 檢出限
取6份空白水樣(4 L),其中3份分別添加不同量的混合標準溶液,配制成含21種目標物質量濃度為0.001,0.002,0.005μg·L-1待測液,另3份空白水樣作空白比對,按試驗方法進行測定,按3倍信噪比計算檢出限(3S/N),結果見表4,同時和氣相色譜-質譜法(GC-MS)所得的檢出限作比對,結果見表4。
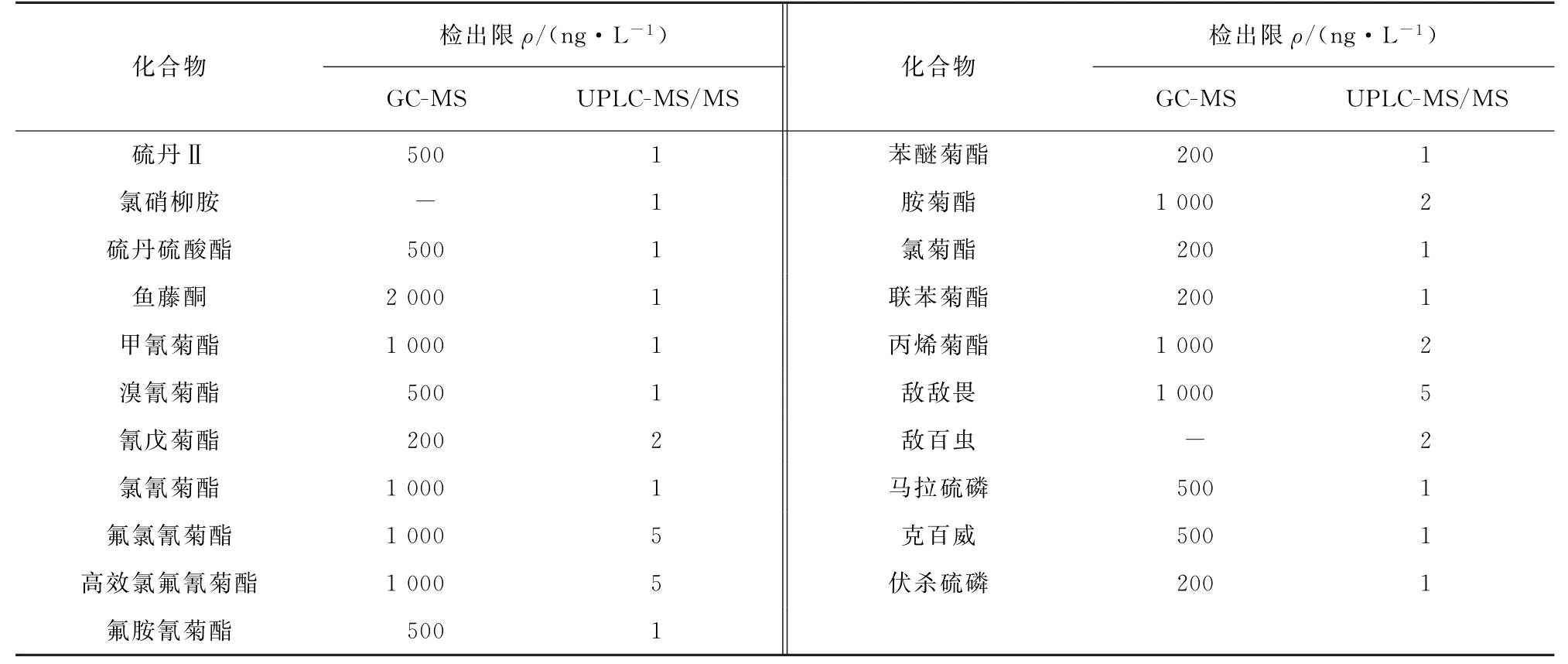
表4 GC-MS和UPLC-MS/MS對21種農藥的檢出限Tab.4 Detection limits of 21 common pesticides found by GC-MS and UPLC-MS/MS
由表4 結果表明:新型吸附型-固相萃取-UPLC-MS/MS 測定水中21 種農藥檢出限為1~5 ng·L-1,遠低于GC-MS的檢出限。實際魚塘投毒案件中的毒物檢驗,因目標物的不確定性,液相色譜-質譜法與氣相色譜-質譜法各具優勢[13],建議二種方法同時使用。
2.6 精密度和準確度試驗
按試驗方法對空白水樣(4 L)進行加標回收試驗,分別添加低、中、高等3個濃度水平的混合標準溶液,配制成質量濃度分別為0.025,0.05,0.5μg·L-1的混合標準溶液各6份。以測定值的相對標準偏差(RSD)考察日內精密度;以連續測定5 d的結果的RSD 考察日間精密度,以加標的每種目標物測得的質量濃度的平均值與此目標物的理論質量濃度的百分比考察方法的準確度[14-15],精密度和準確度試驗結果見表5。
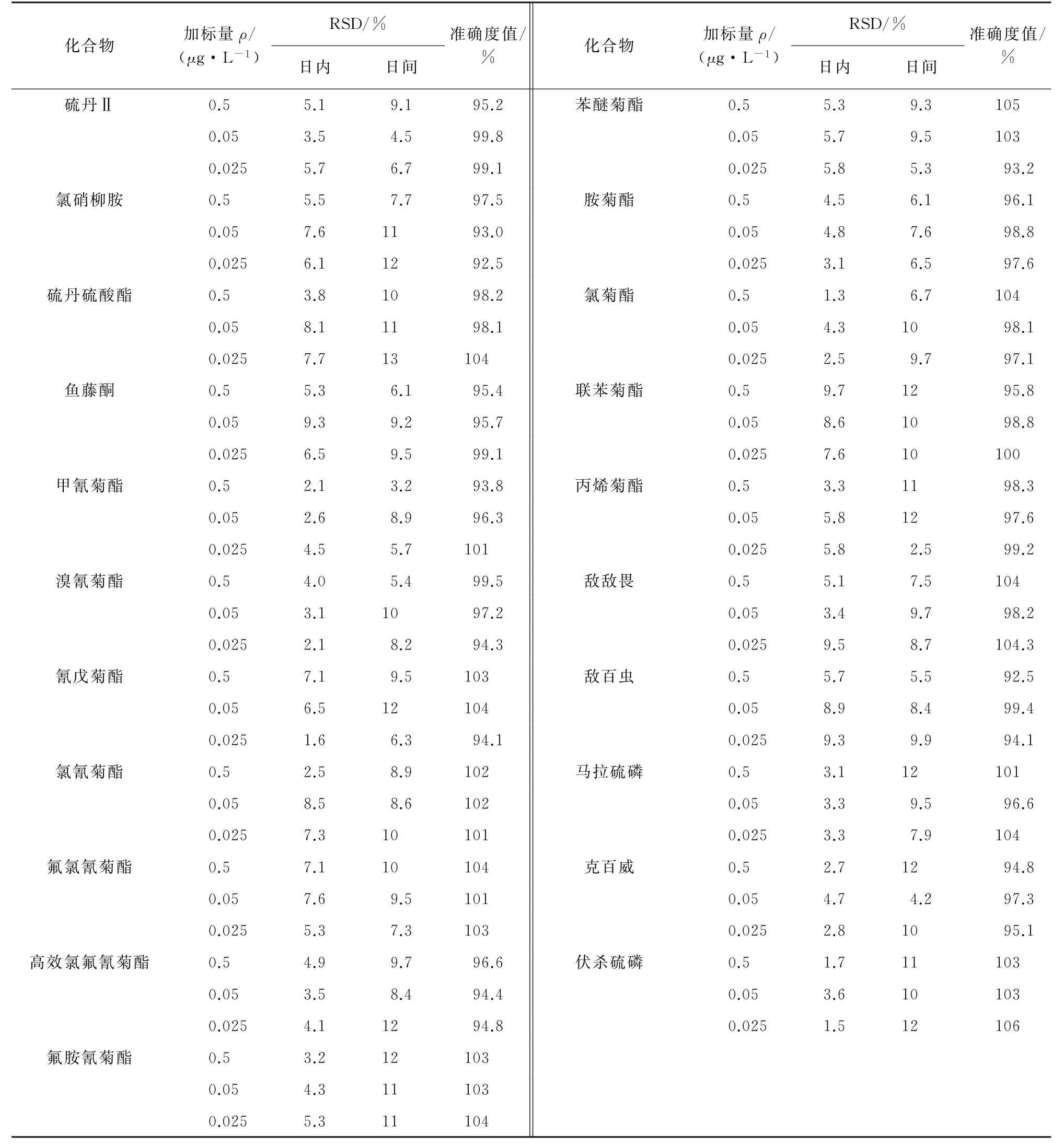
表5 21種農藥的精密度和準確度試驗結果(n=6)Tab.5 Results of tests for precision and accurary of the 21 common pesticides(n=6)
由表5 可知:21 種常見農藥的日內RSD 為1.3%~9.7%,日間 RSD 為2.5%~13%,準確度試驗結果為92.5%~106%。
2.7 樣品分析
2018年12月,揚州某地區發生一起魚塘內魚大量死亡的案件,用本方法從送檢的魚塘水中檢出了農藥氯硝柳胺成分。經查,本案件系事主誤將主成分為氯硝柳胺清塘劑投入魚塘中所致。
2019年2月,某公安分局接到一起魚塘疑似投毒案件,用本方法從送檢的魚塘水中檢出了農藥氯氰菊酯成分,其質量濃度為0.44μg·L-1,經查,本案件是由嫌疑人投放農藥氯氰菊酯所致,檢測出魚塘水中氯氰菊酯的含量與根據嫌疑人交代的投放氯氰菊酯的量和魚塘面積估計出的基本一致。
本工作采用的前處理方法新型吸附劑-固相萃取具有一定創新性,但實際應用中還有許多待改進之處,如不借助任何儀器設備就能快速地將固相填料從大體積水中分離出來,這將是固相萃取-填料薈萃法得到廣泛應用的關鍵。
