MHA實驗室檢查特征2例報道及文獻復習*
2020-04-20 06:01:58李衛濱
國際檢驗醫學雜志 2020年7期
商 蕾,陳 婭,李衛濱
(廈門大學附屬東方醫院/聯勤保障部隊第九〇〇醫院檢驗科,福建福州 350025)
May-Hegglin異常(MHA)是一種少見的常染色體顯性遺傳疾病,該基因定位于染色體22q12.3-13.1[1]。1960年首次用MHA命名此病,多數患者表現血小板減少,伴隨不同程度的出血傾向,少數病例有代謝異常,聽力、視力及腎臟損傷。目前該病發病率超過1/100 000。現分析2例MHA患者病例資料并回顧分析相關文獻,提高臨床和實驗室對該病的認識。
1 臨床資料
本院于2009-2018年確診2例MHA患者,行血常規檢查(ADVIA 2120i,德國)及血涂片鏡檢。經患者簽字同意,行骨髓鏡檢,觀察巨大血小板,并計算粒細胞包涵體百分比。
患者1,女,17歲,2009年11月因血小板進行性減少入院。自述8或9歲開始出現聽力下降。既往2009年7月因“尿毒癥”于本院行同種異體腎移植術。體格檢查:全身皮膚黏膜正常,無皮疹、皮下出血、全身淺表淋巴結無腫大及壓痛。胸骨無叩痛。無視力障礙,雙耳聽力障礙,有尿毒癥。血常規為白細胞計數(WBC) 7.02×109/L;紅細胞(RBC) 3.2×1012/L;血紅蛋白(Hb) 102 g/L;血小板16×109/L。骨髓涂片鏡檢見圖1,示約94%中性粒細胞的細胞質可見藍色包涵體(2~5 μm大小),多位于胞質邊緣,嗜酸和單核細胞中亦可見。大血小板(直徑4~7 μm)占血小板的21%。
患者2,女,45歲,2018年7月因體檢發現血小板減少入院。體格檢查:全身皮膚黏膜正常,無皮疹、皮下出血、全身淺表淋巴結無腫大及壓痛。胸骨無叩痛。無視力障礙,一側聽力障礙,無腎功能異常。自述在十幾歲時一側耳朵因化膿性中耳炎導致聽力喪失。血常規為WBC 5.43×109/L;血小板62×109/L;Hb 142 g/L,RBC 4.87×1012/L。平均血小板體積(MPV) 16.4 fL。血涂片:約88%中性粒細胞的胞質可見有藍色包涵體(2~5 μm大小),嗜酸和單核細胞中亦可見。血小板大小不等,大血小板(直徑4~7 μm)占血小板11%,巨大血小板(直徑>7 μm)占1%。骨髓涂片(見圖2),約87%中性粒細胞的胞質可見藍色包涵體(2~5 μm大小),血小板大小不等,大血小板占血小板 15%,巨大血小板占2%。髂前上棘骨穿病理結果提示造血組織增生稍減低,骨髓腔內見部分巨核細胞呈裸核樣改變,部分巨核細胞分葉減少,未見明確腫瘤樣病變。
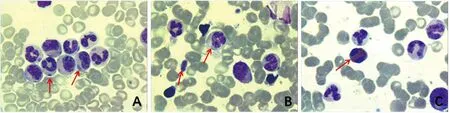
注:A中箭頭表示中性粒細胞藍色包涵體;B中箭頭表示大血小板和中性粒細胞藍色包涵體;C中箭頭表示嗜酸性粒細胞藍色包涵體。
圖1患者1血細胞內藍色包涵體(瑞氏-吉姆薩混合染色法,×1 000)

注:A中箭頭表示中性粒細胞藍色包涵體;B中箭頭表示巨大血小板藍色包涵體;C中箭頭表示嗜酸性粒細胞藍色包涵體;D中箭頭表示單核細胞藍色包涵體。
圖2患者2血細胞內藍色包涵體(瑞氏-吉姆薩混合染色法,×1 000)
2 結 果
進一步進行家系調查結果:患者1,對其有血緣關系的親屬(父親、母親、兄長、姐姐),進行了血常規及血涂片觀察,未見血小板減少、巨大血小板、中性粒細胞包涵體的存在,家屬拒絕行基因檢測。可初步排除該家系5個成員患有MHA。患者2,對其有血緣關系的家庭成員(姐姐、妹妹、兒子)進行了血常規及血涂片觀察,其中,姐姐有輕微貧血,但無其他異常,三者均無血小板減少、巨大血小板、中性粒細胞包涵體的存在,家屬拒絕行基因檢測,故初步排除MHA可能。兩例均可能為散發病例。
3 討 論
MYH9基因(MYH9)編碼非肌性肌球蛋白重鏈Ⅱ(NMMHC-Ⅱ),其異常常導致MHA、Fechtner綜合征(FTNS)、Epstein綜合征(EPS)和Sebastian綜合征(SBS) 等疾病。典型病例具有巨大血小板、血小板減少和中性粒細胞包涵體“三聯征”。如本文新報道的2例MHA患者。應與Alport綜合征鑒別,兩種疾病均可表現視力、聽力、腎功能受損,但后者無粒細胞系包涵體。
研究顯示MHA患者中性粒細胞異常藍色包涵體和NMMHC-Ⅱ相關,后者包括NMMHC-ⅡA,NMMHC-ⅡB和NMMHC-ⅡC。 NMMHC-Ⅱ A表達于粒細胞系、淋巴細胞系及巨核細胞系中,血細胞中藍色包涵體和NMMHC-ⅡA異常聚集、組裝導致細胞骨架成分改變相關[2]。在本院發現的2例病例中首次獲得在血小板及嗜酸細胞中藍色包涵體的圖片。MHA藍色包涵體和杜勒氏小體鑒別:前者較大且界限清楚,后者常呈圓形界限不清的云霧狀;出現杜勒氏小體時,常伴有中性粒細胞中毒改變及感染等臨床癥狀,一般無巨大血小板的出現,感染控制后杜勒氏小體消失;MHA患者藍色包涵體為終身存在。在臨床診斷中主要使用瑞吉染色初篩,MHA必要時可使用電鏡檢查及免疫熒光技術。血小板NMMHC-ⅡA可分解ATP產生能量、牽動肌動蛋白,其異常可導致血小板骨架異常,促進未成熟血小板釋放入血,導致血小板減少及巨大血小板。大血小板會造成血小板計數假性減低,為避免漏診,本院建議首次低于80×109者,應進行血涂片復檢[3]。此外,MHA患者血小板計數減少需與特發性血小板減少性紫癜(ITP)鑒別,ITP一般MPV值正常,而MHA的MPV值偏大;ITP鏡檢中性粒細胞無藍色包涵體,一般有巨核細胞增多伴成熟障礙,且80% ITP患者可檢出相關抗體。
回顧目前國內報道病例,共24人,男性患者7人,女性患者16人,5個家系報道[4-7]。其中13人行基因檢測,其報道突變位點分別為第17號外顯子G2105A、第25號外顯子c.3195_c.3215 delCGA GCT CCA GCC CAG ATC GC,T3429G、第30號外顯子G/A突變致氨基酸突變D1424N、第38號外顯子G5701A、G5521A、第40號外顯子T2256C,T3429G、C5797T、5803delG。2011年意大利科學家分析218例MYH9-RD不相關家系,79%的患者突變只影響6個氨基酸殘基位點,球狀頭部的Ser 96(6%)、Arg 702(24%),螺旋結構域的Arg 1165(9%)、Asp 1424(20%)和Glu 1841(22%),或尾部螺旋狀結構域的Arg 1933(19%)[8-9],目前已經發現了80個家族基因突變位點[10]。其中頭部基因域的突變更易導致嚴重的巨大血小板減少、早期腎病、感音神經性耳聾,但是粒細胞藍色包涵體有時不可見[11]。MYH9-RD有35%的散發病例,國內該病例多以散發形式報道,常提示新的突變位點。但是隨著此種疾病報道增多,發現不僅相同錯義突變可導致不同的病情,同一同義突變也可導致疾病的輕重緩急,而確診MHA患者也可能基因測序結果未見異常,這提示MYH9基因也許不是致病的唯一基因,其中關系還需要進一步探索。許多MHA患者常患有其他疾病,例如貧血[12]、血液及其他系統惡性腫瘤[13-15]。
國內外文獻中提及因誤診使用皮質激素、大量免疫球蛋白及脾切除等治療對MHA減輕出血及升高血小板均無效,但輸注去氨加壓素或應用一種人工合成的抗利尿劑衍生物(1-去氨基-8-右旋-精氨酸-加壓素,DDAVP)能明顯縮短患者出血時間。最近的文獻提示使用一種口服非肽類血小板生成素受體激動劑(艾曲波帕)可明顯改善血小板減低[16-17]。根據臨床經驗,長期服用維生素C,可起到一定的預防作用。對于MYH9基因突變導致的腎病綜合征的治療,動物實驗研究顯示使用坎地沙坦可減輕慢性腎病的進程[18],但是臨床試驗還在進一步完善中。
