鉑催化硅氫加成反應研究進展
2020-04-01 10:04:30柯其寧代志鵬陳琛陳緒煌
化工進展 2020年3期
柯其寧,代志鵬,陳琛,陳緒煌
(湖北工業大學綠色輕工材料湖北省重點實驗室,湖北武漢430068)
鉑催化劑是硅氫加成反應目前最常用的催化劑,有著廣泛的研究。硅氫加成反應作為合成有機硅化合物最重要的途徑之一,為當今的研究熱點與難點。自Sommer等[1]發現硅氫加成反應以來,大量文獻對其進行了報道[2]。以鉑等金屬及其鹽作為硅氫加成的催化劑最早在一些專利中就有提及,Wagner[3]首先對鉑作為催化劑進行了詳細的研究。1957 年,Speier 等[4]發現氯鉑酸催化劑,即氯鉑酸的異丙醇溶液對硅氫加成反應具有較高的催化活性和催化效果。但其有催化劑用量大、催化選擇性較低、催化范圍較小、合成過程產生氯等缺點。1973年,Karstedt[5]發現了一種具有高催化活性的鉑的乙烯基硅氧烷配合物,即Karstedt 催化劑。相比Speier催化劑,其催化活性更好、用量更少、應用范圍更廣、合成過程無氯產生。但兩者均存在回收困難、成本較高、催化難以控制、催化選擇性不夠優良的缺點,導致了目前工業上鉑損失嚴重。隨著資源的日益匱乏,可回收的負載鉑催化劑[6-9]將成為硅氫加成反應研究中的一個重要的新領域。負載鉑催化劑具有可回收、成本低、催化選擇性較好的優點。其在催化反應后可通過過濾、離心等手段進行分離,可多次利用;負載催化劑用量一般相對較少且重復使用次數較多,明顯降低了成本;其催化活性點一般可與反應物直接接觸,可直接進行催化,催化選擇性相對較好。目前主要以無機負載鉑催化劑研究最廣,有機類、固載液鉑催化劑以及其他新型催化劑也有文獻報道。
本文主要綜述了近年來硅氫加成反應均相鉑催化劑以及負載化鉑催化劑的研究進展,并對其催化性能和特點進行了評價總結。
1 硅氫加成反應機理
對于鉑催化硅氫加成反應機理,目前尚未有明確的定論,其具體的機理仍有待研究,普遍接受的機理主要為兩種:Chalk-Harrod 機理及膠體鉑機理。1965年,在對氯鉑酸的研究基礎上,Chalk 和Harrod[10]通過研究鉑催化硅氫加成反應發現:鉑催化劑的活性來自其金屬中心([M]),首先Si H 鍵斷裂后對鉑金屬中心進行氧化加成,形成過渡態的Si [M] H結構;而鉑中心的配位作用使得雙鍵變弱,具有加成的傾向;隨后發生硅氫加成反應,最后還原消除,重新生成活性中心[M]。鉑金屬催化的硅氫加成反應可以由Chalk-Harrod機理總結,如圖1所示。
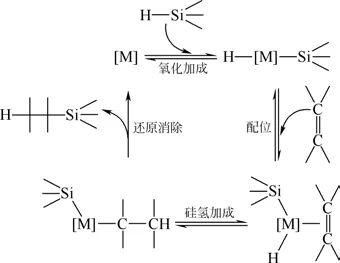
圖1 過渡金屬催化硅氫加成Chalk-Harrod機理[10]
但Chalk-Harrod機理無法解釋一些硅氫加成反應產物具體質量的計算和反應液泛黃的現象。隨后,Larry 在電鏡下發現了膠體鉑的存在,Sakaki等[11]在此基礎上對之前的Chalk-Harrod 機理提出了修正,即膠體鉑機理,其催化反應過程如圖2所示。Chalk-Harrod 機理認為烯烴的雙鍵插入[Pt]IIH中;而膠體鉑機理認為烯烴的雙鍵也有部分插入到[Pt]II-SiR3上。兩種過程同時發生,最后還原消除,釋放出產物。

圖2 膠體鉑機理圖[11]
2 均相鉑催化劑
目前工業化的均相鉑催化劑以Karstedt 催化劑為主,其缺點是催化過程不易控制、催化選擇性低、基本無法重復使用,提高均相鉑催化劑的選擇性和催化范圍顯得十分必要。由于成本和反應機理不夠深入的限制,關于均相鉑催化劑的研究還相對較少,本文對含有多個鉑原子的鉑簇化合物催化劑、N-雜環卡賓鉑配合物催化劑和聚合物鉑配合物催化劑進行了報道分析。
2.1 鉑簇化合物催化劑
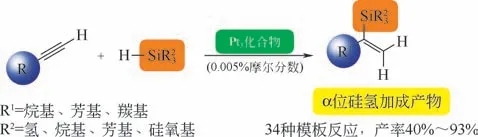
圖3 Pt3簇化合物催化劑[12]
Karstedt 催化劑催化產物一般為β加成產物,而在某些硅氫加成反應中,α位加成產物才具有商業價值。Corma 等[12]合成了3 種Pt3簇化合物催化劑[NEt4]2[Pt3(CO)6]3、Na2[Pt3(CO)6]5和Na2[Pt3(CO)6]10,該類催化劑可定向催化合成α位產物,通過研究分析34 種烴基、炔基等的硅氫加成反應,如圖3 所示,相比于傳統Karstedt 催化劑幾乎無α位的催化選擇性,其α加成產物產率為40%~93%,且轉化效率(TOF)極高,達106h-1。其他關于定向催化生成α位加成產物研究未見報道,該項研究成果具有創新性,改善了均相鉑催化劑的催化選擇性。其缺點是其添加量較多(50μg/g),催化選擇性還有待提高。
2.2 N-雜環卡賓鉑配合物催化劑
N-雜環卡賓配合物(NHC)由于物理化學性質穩定,具有一定的空間性能,逐漸在催化加成反應中得到應用[13-15],尤其是烯、炔烴的硅氫加成反應,催化效果良好,是均相鉑催化劑的理想配體。Zhang等[16]合成了硅基功能化的含有聚醚鏈的N-雜環卡賓鉑配合物催化劑(mPEG-NHC-Pt),其具有極佳的催化選擇性和重復使用性。在模板烯烴加成反應中催化選擇性達99.7%,炔烴加成反應達99.8%。通過離心上層液體后,催化劑可重復使用27 次,原因是鉑與N-雜環卡賓配合物鍵合力十分牢固,催化過程中鉑幾乎未發生損失。遺憾的是,目前其具體的催化機理仍在研究之中,其生產成本也較為昂貴。
2.3 聚合物鉑配合物催化劑
將Pt 引入到聚合物鏈中,通過鏈段的長短可以有效地控制Pt 濃度,從而更好地控制硅氫加成反應催化過程。機械化學反應是一種利用機械能誘發化學反應或誘導材料結構和性能發生變化來制備新材料的方法,利用該方法所得的聚合物稱為機械反應性聚合物。Wei 等[17]通過聚合物機械化學反應合成了一種Pt 兩端含乙炔基的聚合物催化劑,合成過程如圖4所示。
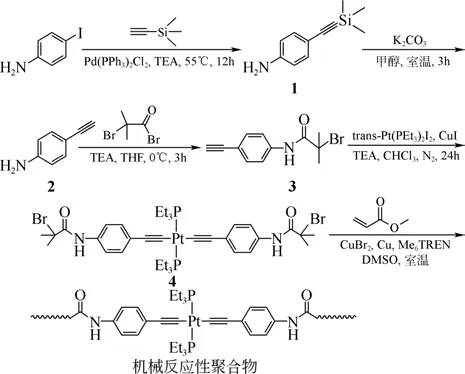
圖4 鉑-乙炔基絡合物的機械反應性聚合物的合成[17]
首先通過4-碘苯胺與(三甲基)乙炔在一定條件下反應堿洗后得到中間體2;然后與甲基丙酰溴反應得到化合物3;再通過催化脫氫鹵化反應得到引發劑4;最后利用自由基聚合得到機械致敏聚合物催化劑,即其催化活性受外加機械作用影響較大的催化劑。該催化劑在超聲誘導的條件下才會發生離解,暴露出的鉑為催化活性點,催化硅氫加成應。在沒有機械誘導的條件下幾乎無催化效果,可實現對催化過程的控制。缺點是其催化活性還不夠高,與活性鉑的離解程度及聚合物Pt 催化劑的分子鏈長度有關。催化反應的控制是催化反應的關鍵問題之一,這類通過外加機械作用的變化調控催化劑的活性,利用機械手段控制硅氫加成反應,具有重要的研究價值。
3 負載鉑催化劑
目前均相鉑催化劑催化選擇性不高,難以分離回收。減少鉑的損失,降低成本,提高催化劑的使用效率,是新一代負載鉑催化劑的研究方向。目前已有大量研究進行了深入報道[18-20],但大部分處于實驗室研究階段,商用催化劑種類較少,進一步的工業化還有待深入。
3.1 SiO2負載鉑催化劑
SiO2鉑催化劑負載體一般為硅膠,表面含有活性基團,為鉑催化劑的優良載體。其化學性質穩定,幾乎不溶于水和任何溶劑,制作成本低,具有一定的機械強度、孔隙結構及吸附能力。基于以上優點,其適合作為某些金屬及金屬鹽的載體,受到了廣泛的關注[21-23]。
Li 等[24]在之前學者[25]的研究基礎上,將乙二胺四乙酸(EDTA)接枝到含有氨丙基三乙氧基硅烷的硅膠載體上,制備了一種新型SiO2負載鉑催化劑SiO2-EDTA-Pt,在模板加成反應中,其催化產物均為β型,選擇性極強,經多次回收使用后,催化產率仍在80%以上,催化性能大幅改善,但其分析僅涉及單個模板反應。為探索多種催化反應的催化性能,Hu 及Prinsen 等[26]合成了一種超微孔硅負載鉑催化劑,鉑的負載量為0.7%,孔體積在0.7~0.9cm3/g,孔徑小于2nm,符合超微孔材料的特征,研究了反應物配比、溫度對催化劑的選擇性和活性的影響。所制催化劑可與不同烯炔烴(乙炔、辛烯等)硅氫加成反應,經7次使用后,產物和β加成產物轉換率均超過97%,催化選擇性和重復使用性尚佳。且該催化劑用丙酮和乙醇洗滌后,可較易從反應介質中分離。
3.2 碳負載鉑催化劑
碳負載鉑催化劑載體以活性炭應用最廣,氧化石墨和石墨烯也有涉及。活性炭由于其豐富的孔內表面、不飽和價以及缺陷位,因而表面可充分負載催化劑催化活性中心。自Wagner[3]首次報道了以活性炭為固載材質制得Pt/AC催化劑后(圖5為Pt/AC催化硅氫加成反應的反應式),越來越多的研究者致力于活性炭負載鉑催化劑在硅氫加成反應上的研究,某些產品已經實現工業化,近年來發展方向逐步轉向催化劑催化能力的影響因素的探究[27]。
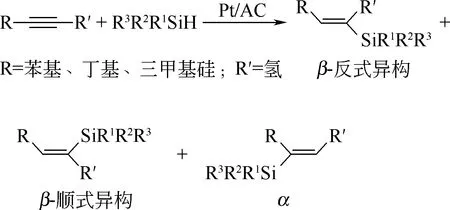
圖5 Pt/AC催化硅氫加成反應[3]
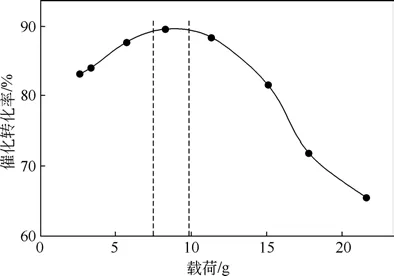
圖6 不同負載下硅氫加成反應轉換率[28]
Dorokhov 等[28]在不同接觸載荷條件下分析了碳負載催化劑的催化性能變化情況,結果如圖6 所示,縱坐標為催化轉化率,橫坐標為平均每克催化劑所受載荷大小,發現負載過大或者過小都會影響催化性能,轉化率最高超過90%,而最低低至70%以下。其原因與反應物有關,也表明了鉑負載能力對催化效果的影響,同時活性炭負載催化劑的內部改性是提升其催化能力的一個關鍵因素,決定著活性炭負載催化劑的催化性能。
氧化石墨可修飾具有活性基團的有機高分子,其活性基團與氧化石墨嫁接[29],而其他基團則與鉑形成配合物[30-32]。Rao 等[33]利用乙烯基三乙氧基硅烷將鉑固定在氧化石墨表面,合成了一種類Karstedt 催化劑結構的氧化石墨烯(GO)-Karstedt催化劑,如圖7所示。該催化劑催化己烯與三乙氧基硅烷反應的β加成產物的產率達92.5%,且反應物完全轉化。經數次回收使用后,轉化率及β加成產物產率均無明顯下降。但由于GO負載鉑催化劑合成產率的影響及接枝基團的不確定性,其催化活性和選擇性相比于三維的負載催化劑較低,同時關于GO表面其他基團修飾的研究也有待發掘。

圖7 GO-Karstedt催化劑的合成[33]
石墨烯表面經過化學處理后可與催化活性組分發生鍵合作用,也可用來負載鉑催化劑。Kong等[34]合成了一種石墨烯納米板(GNP)負載鉑催化劑Pt-GNP,其催化轉化效率(TON)為9.4×106,遠大于Karstedt催化劑的0.9×106,表現出良好的催化活性。催化烯烴與硅烷的硅氫反應,如圖8 所示,縱坐標分別為產物轉化率和Pt 損失率,其5h 內仍可保持90%的轉化率。8h 后用環戊基甲基醚(CPME)洗滌,其催化活性又恢復到80%左右,表現出良好的催化穩定性。
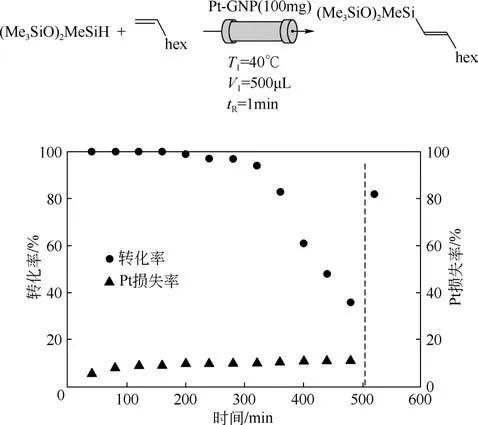
圖8 Pt-GNP催化烯烴加成反應[34]
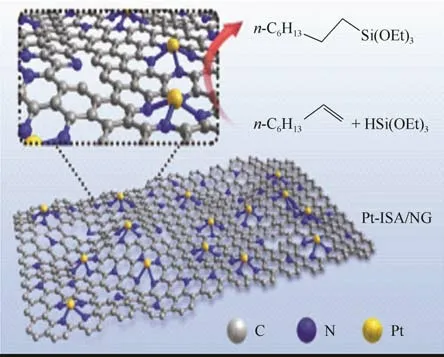
圖9 Pt-ISA/NG催化劑的結構示意圖[36]
以上碳負載催化劑選取的催化模板反應都較少,Zhu 等[35]進一步擴展研究了催化范圍,通過Na2CO3輔助一鍋熱解法[36]合成了一種氮摻雜的石墨烯(NG)鉑單原子位點(ISA)二維結構的多相負載催化劑Pt-ISA/NG,如圖9 所示,具有超高比表面積和高金屬負載密度的特點。以三乙氧基硅烷與多種烯烴的硅氫加成催化反應為模板反應,結果顯示均具有較好的選擇性,反馬氏加成產物均在98%以上,轉化率均在92%以上,催化活性較高。以己烯為模板反應,其5次催化使用后仍可達到94%的催化轉化率。無論催化活性還是催化選擇性都比目前商用的碳負載催化劑要高,具有工業化應用的潛力。缺點是由于異相催化劑存在空間效應[37],隨著烯烴分子量和催化劑尺寸變大,催化效果變差,而Pt-ISA/NG只是微米級別的二維分布,催化劑尺寸和分散的穩定性也需要較好地控制。
3.3 金屬氧化物負載催化劑
納米金屬氧化物具有極大的比表面積和極高的化學穩定性,且與鉑之間的作用力較強,是理想的鉑催化劑載體之一。
Cui和Junge等[38]采用浸漬法制備了一種Pt單原子Al2O3納米棒負載催化劑Pt/NR-Al2O3-IP,通過與不同烯烴、不同叔代硅烷、不同聚硅氧烷等的催化反應,表明其具有極高的催化選擇性和較廣的催化范圍;重復使用6次后催化產率仍達92%以上,重復使用性優良。在以辛烯與三乙氧基硅烷為模板反應時,其催化活性與商用Karstedt 催化劑接近,催化活性較高。
由于Pt 單原子催化劑(SACs)具有良好的金屬分散性和最大效率[39-40]以及層狀金屬氧化物特性[41],Chen等[42]利用靜電感應離子交換技術和二維約束方法合成單原子TiO2負載Pt 催化劑,合成過程如圖10 所示。該催化劑具有良好的催化活性和選擇性,通過不同烯烴與不同硅氧烷的硅氫加成催化反應,其催化轉化率為97%~99%,產物選擇率在90%~97%,有望實現工業化。
3.4 有機高分子負載鉑催化劑
有機高分子由于結構易被修飾,成為負載鉑的良好載體。目前除了傳統的聚酰胺類鉑催化劑外,聚硅氧烷類、金屬有機骨架類等聚合物負載鉑催化劑也有研究。
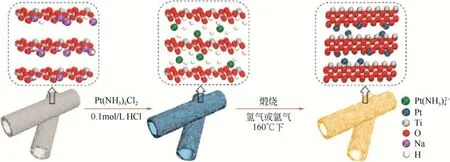
圖10 Pt1δ+/TiO2合成過程[42]
聚酰胺類聚合物是最早用來作為鉑催化載體的有機聚合物載體之一,其含有與鉑可配位的酰胺基團,易形成鉑負載催化劑。Michalska等[43]以含有吡啶的聚酰胺為載體,合成了一種Pt 負載催化劑,在苯乙炔和三乙氧基硅烷的硅氫加成反應中具有較好的催化選擇性,并驗證了其催化選擇性與引入的供電子基團的強弱相關。但其催化活性和重復使用性都不夠高,催化范圍也較小,催化性能有待提升。
聚硅氧烷類高分子具有易調控的鏈式結構,通過改變結構可使催化的反應物與活性中心更易結合,是非常理想的鉑催化載體。Chauhan 等[44]合成了一種交聯聚硅氧烷負載的鉑催化劑,如圖11 所示,由二甲基環烯烴的鉑配合物與2000 左右分子量的聚硅氧烷在溫和條件下即可合成,透射電鏡結果顯示鉑納米粒子分布在聚硅氧烷的交聯結構中。其在三乙氧基硅烷、三甲基硅氧烷-1-丙基、苯乙炔和二甲基氯硅烷的硅氫化反應中具有極佳的催化活性和選擇性,大多數反應產物選擇率為100%,轉化率均在95%以上。該催化劑制備條件溫和、易操作、催化活性高、成本較低,但其催化范圍需要進一步擴大,距離工業化生產還有一定難度。
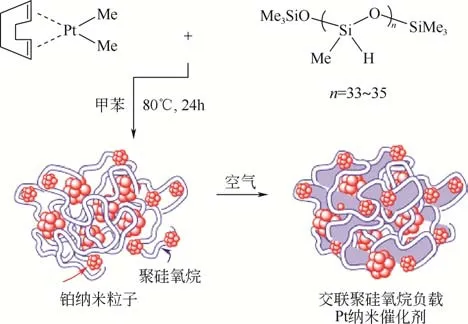
圖11 交聯聚硅氧烷負載Pt納米催化劑的合成[44]
近年來,金屬有機骨架類載體因其相對無機載體具有更大的比表面積和更為可控的孔結構等優點,成為一種合適的催化劑載體[7]。謝志凱等[45]利用浸漬法制備了一種硅烷偶聯劑改性的含鉻金屬的有機骨架鉑負載催化劑。其對多種烯烴聚醚具有良好的催化活性,催化轉化率最高達94%,5次循環使用后仍可達到86%以上,反應條件溫和,90℃下便可迅速反應。缺點是其對高分子量的催化底物催化活性較低,轉化率不足68%,這與催化劑和底物的相容性有關;也受到催化劑純度的影響,同時催化劑用量較多,為72μL/L。
3.5 固載液鉑催化劑
離子液體作為一種新型的催化劑載體已得到一定的應用[46-47]。固載液催化劑在室溫下為固體,易與催化體系分離,溫度較高時為液體,相容性好,催化效率高,可多次重復使用。Magdalena 等[48]探究了5種不同的嗎啉離子溶液負載的鉑催化劑的催化性能,其催化轉化率最高可達95%,均高于80%,重復使用10 次后活性無明顯下降,但未涉及催化選擇性的分析。在此基礎上,Fernández等[49]采用乙二醇/氫氧化鈉混合液在160℃下還原氯鉑酸,然后在室溫下負載于四氟硼酸三咪唑溶液(L2)后溶于四氫呋喃,制備出了可溶性納米鉑固載液催化劑(Pt/NPs),合成示意圖如圖12 所示,EG為乙二醇,在高溫堿溶液下還原氯鉑酸,L2為Pt納米粒子的穩定劑,可提高其負載能力。各種炔烴的硅氫加成反應的研究結果表明其催化選擇性和活性均較高,回收5 次使用后催化活性無明顯降低。不足的是,催化過程均有鉑納米粒子的損失,鉑的負載能力有待增強,且其催化的區域選擇性僅僅是對一些內炔和不對稱炔烴較好,限制了其應用與發展。
3.6 其他新型鉑負載催化劑
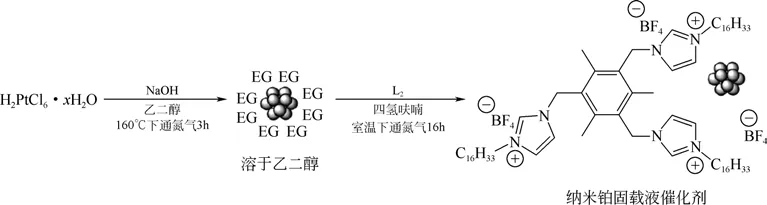
圖12 納米鉑固載液催化劑的制備[49]
除上述催化劑外,一些新型的鉑負載催化劑也有文獻報道。劉國旺[50]合成了一種以殼聚糖為配體(CS)埃洛石納米管(HNTs)負載的鉑催化劑Pt-CS/HNTs,催化反應5h后轉化率可達80%,產物選擇率達90%,5次分離回收重復使用后催化活性無明顯下降。該催化劑催化范圍較小,轉化率較低,但催化選擇性較高,且埃洛石廉價易得,符合目前鉑催化劑的發展趨勢。陳偉等[51]制備了粒徑為2~4μm 的SBA-15微球分子篩負載型鉑催化劑,催化轉化率雖僅有68.2%,5 次使用后轉換率為62.3%,與其孔徑小、孔壁薄、化學穩定性差相關,但其解決了負載鉑催化劑分散性問題,并具有貫通介孔結構和高比表面積,催化劑用量也明顯減少。為解決負載催化劑分離回收復雜的問題,含有磁性材料的負載催化劑成為領域熱點。具有超順磁性的磁性材料載體使鉑負載催化劑在外加磁場條件下就能輕易分離,回收效率和利用率均較高[52]。Zai等[53]合成了一種磁性納米鉑催化劑Pt/SiO2/Fe3O4,經重復回收7次后,催化產率仍未發生明顯下降,鉑的負載能力良好,分離簡單,成本明顯降低,具有工業化應用價值。
4 結語及展望
近年來,均相及負載鉑硅氫加成反應研究活躍,取得了顯著的成果。均相鉑催化劑致力于提高其催化選擇性、控制其催化過程、實現均相催化體系的分離回收。負載鉑催化劑為當前鉑催化領域的研究重點,其高催化選擇性、重復使用性、可回收性均是目前所需催化劑的特性,同時催化活性和鉑的負載能力是不可忽視的問題。催化載體以無機碳和SiO2最多,未來有機載體和固載液負載催化劑也具有獨有的發展潛力。對于負載鉑催化劑,載體上接枝氨基、巰基等功能基團是提高其催化選擇性的重要手段之一,也是提高鉑負載能力的有力手段。由于分離過程的復雜和成本問題,利于分離的磁性納米負載鉑催化劑將解決這個問題,其將是未來重要的負載鉑催化載體。催化機理的研究將決定著鉑催化劑的應用前景,但目前成果較少。同時,無論均相還是負載鉑催化劑,其催化反應范圍的問題也是一大熱點,目前僅涉及部分烯、炔烴的硅氫加成反應,催化范圍的擴大化也是一大發展方向。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車工程學報(2017年2期)2017-07-05 08:13:02
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
