伴丘疹性損害的先天性無毛癥一例HR基因突變檢測(cè)
2020-03-16 00:55:00劉偉英高貴云鄭璐瑤
中國(guó)麻風(fēng)皮膚病雜志 2020年1期
劉偉英 高貴云 鄭璐瑤 李 明
1湖南航天醫(yī)院皮膚科,長(zhǎng)沙,410205;2上海交通大學(xué)醫(yī)學(xué)院附屬新華醫(yī)院皮膚科,上海,200092
先天性無毛癥是一組罕見的遺傳性皮膚病,具有較強(qiáng)的臨床和遺傳異質(zhì)性,其臨床表現(xiàn)為頭皮或身體其他部位的毛發(fā)缺失或完全脫光,伴或不伴有身體其他系統(tǒng)的損害,根據(jù)表型可分為綜合征型和非綜合征型。先天性少毛癥的遺傳模式常呈常染色體顯性遺傳、常染色體隱性遺傳以及性連鎖遺傳。伴丘疹性損害的先天性無毛癥(atrichia with papular lesions, APL)的遺傳模式為常染色體隱性遺傳,其致病基因是HR基因[1]。APL患者出生時(shí)毛發(fā)正常,出生后不久頭皮毛發(fā)完全脫失且不可逆,大概2歲時(shí),患者開始出現(xiàn)多發(fā)的毛囊丘疹,出現(xiàn)在面部、頭皮、肢端等,某些患者還可見色素減退斑。本研究中,我們對(duì)一例APL患兒進(jìn)行了HR基因的突變檢測(cè),現(xiàn)將結(jié)果報(bào)道如下。
1 對(duì)象與方法
1.1 對(duì)象 患兒,女,8歲。足月順產(chǎn),出生時(shí)毛發(fā)與胎脂粘連于頭皮,洗澡后頭發(fā)、眉毛、睫毛大部分脫落,持續(xù)加重,頭發(fā)脫落后不生長(zhǎng),眉毛、睫毛可再生長(zhǎng)但不牢固,長(zhǎng)至約1 cm后脫落。1歲時(shí)患兒頭發(fā)脫光,近幾個(gè)月頭皮出現(xiàn)散在近膚色丘疹。父母均正常,無兄弟姐妹,家族中其他成員無類似表現(xiàn)。體檢:患兒血尿糞常規(guī)、肝功能、免疫功能、微量元素測(cè)定均無明顯異常,智力正常。皮膚科檢查:頭發(fā)、眉毛、睫毛全部脫失(圖1a、1b),頭皮可見散在近膚色毛囊丘疹(圖1c),無掌跖角化,無指(趾)甲異常。
1.2 外周血基因組DNA抽取 征得知情同意后,抽取患兒及其父母共3人的外周靜脈血各2 mL。同時(shí)采集了100名正常對(duì)照的外周血。應(yīng)用QIA ampDNA Blood Minikit(德國(guó)QIAGEN公司)試劑盒提取基因組DNA。
1.3 PCR擴(kuò)增和測(cè)序 采用Primer 5.0軟件對(duì)HR基因的19個(gè)外顯子設(shè)計(jì)特異性引物,進(jìn)行常規(guī)PCR擴(kuò)增,PCR擴(kuò)增產(chǎn)物純化后使用ABI 3730測(cè)序儀進(jìn)行Sanger測(cè)序,測(cè)序結(jié)果使用Chromas軟件進(jìn)行分析,并通過與HR基因cDNA參考序列(NM_005144.4)比較來描述突變。
2 結(jié)果
DNA直接測(cè)序結(jié)果顯示HR基因存在復(fù)合雜合突變,分別在第10外顯子發(fā)現(xiàn)移碼突變c.2270delC(圖2a),導(dǎo)致第757位氨基酸提前終止,變?yōu)闊o義氨基酸。在第15外顯子內(nèi)發(fā)現(xiàn)錯(cuò)義突變c.3038T>C(圖2b),導(dǎo)致1013位氨基酸由脯氨酸突變?yōu)榱涟彼帷;純旱膬蓚€(gè)突變分別遺傳于其父親和母親。以上2個(gè)突變?cè)?00名正常對(duì)照中均未檢測(cè)到(圖2c、2d),提示它們?yōu)橹虏∽儺惗菃魏塑账岫鄳B(tài),查閱國(guó)內(nèi)外文獻(xiàn)均屬首次報(bào)道,在Ensemble、NCBI等數(shù)據(jù)庫(kù)均未查到該突變。
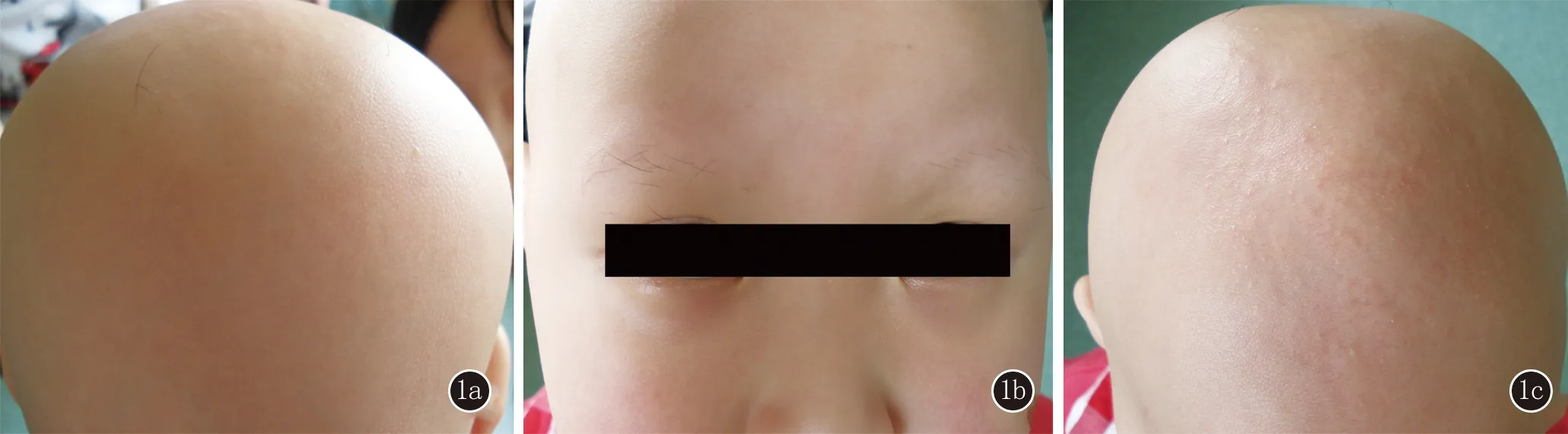
1a、1b:患兒頭發(fā)和眉毛脫失;1c:頭皮可見近膚色的毛囊丘疹
圖1臨床圖片
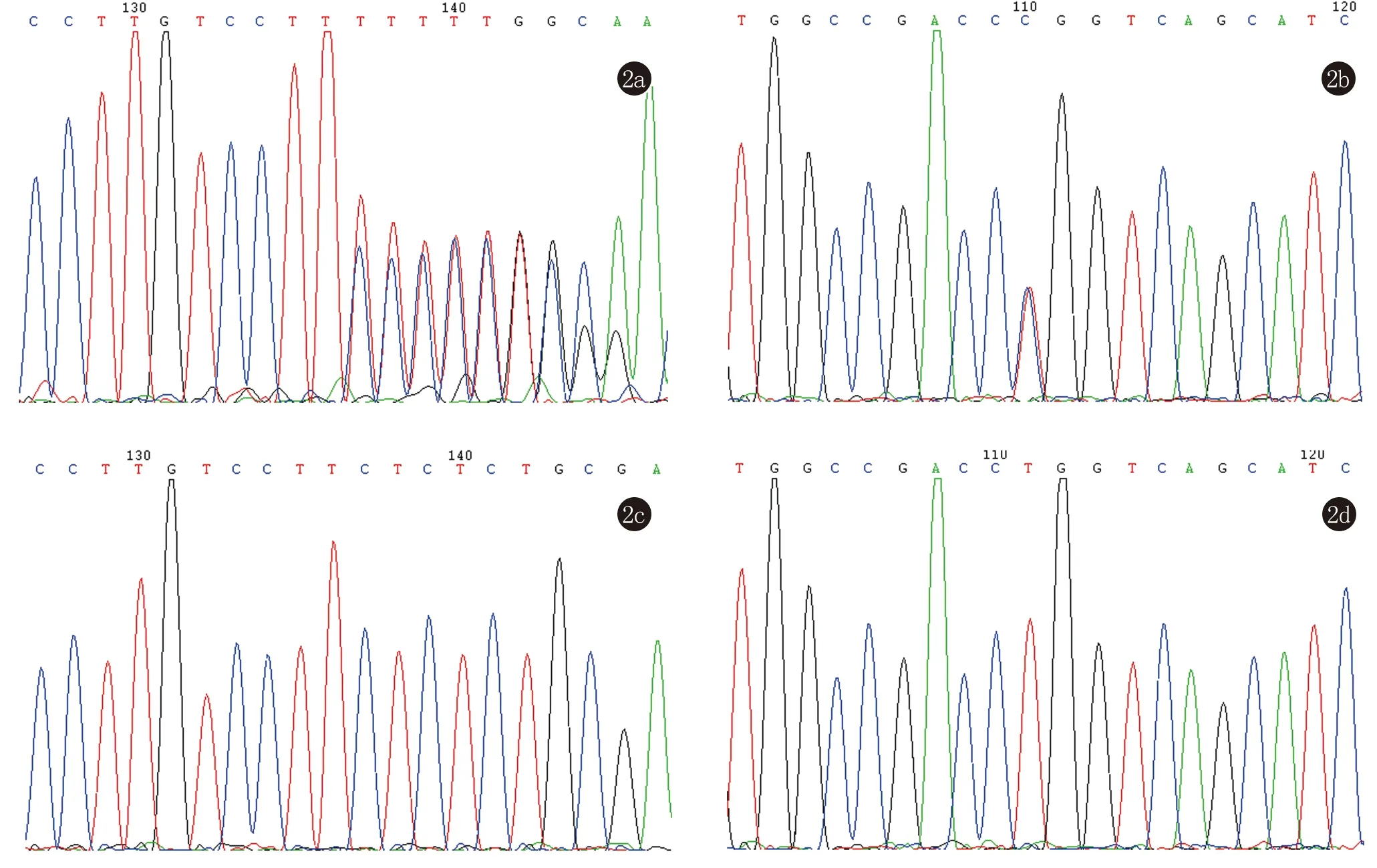
2a:示c.2270delC移碼突變;2b:示c.3038 T>C錯(cuò)義突變;2c:正常對(duì)照序列;2d:正常對(duì)照序列
3 討論
伴丘疹性損害的先天性無毛癥患者臨床上可表現(xiàn)為出生時(shí)局部或全部的毛發(fā)缺失或者出生時(shí)毛發(fā)正常,在出生后的前幾個(gè)月脫落,患者還會(huì)出現(xiàn)擴(kuò)散的多發(fā)性角化毛囊性丘疹,好發(fā)于頭皮、面部和四肢。患者無其他外胚層發(fā)育的缺陷,如指甲、汗腺或牙齒等,某些患者軀干部可見色素減退斑。APL的遺傳模式通常為常染色體隱性遺傳。本文報(bào)道的患兒為散發(fā)病例,出生后不久毛發(fā)均脫落,同時(shí)伴有丘疹損害,為典型的常染色體隱性遺傳APL。
APL通常需要與先天性普禿(alopecia universalis congenita,AUC)進(jìn)行鑒別診斷,兩種疾病均為常染色體隱性遺傳,且病因都與無毛基因的突變有關(guān),值得注意的是,AUC患者表現(xiàn)的毛發(fā)脫失和APL幾乎一致,但前者皮膚是正常的,而APL患者會(huì)出現(xiàn)擴(kuò)散的多角化毛囊性丘疹。仔細(xì)詢問家族史,根據(jù)典型的臨床表現(xiàn)不難作出診斷。
1954年,Damste等[2]首次報(bào)道了APL,1998年Ahmad等[3]在一個(gè)以色列-阿拉伯裔的家系中將該病的致病基因定位于染色體8p21[4],并發(fā)現(xiàn)HR基因?yàn)楸静〉闹虏』颍摶蚝?9個(gè)外顯子,只表達(dá)在皮膚和大腦,其表達(dá)產(chǎn)物是無毛蛋白,分子量為130kDa,含1189個(gè)氨基酸,該蛋白功能的缺陷會(huì)導(dǎo)致毛發(fā)生長(zhǎng)周期的不穩(wěn)定,其可能在第一個(gè)毛發(fā)生長(zhǎng)周期起作用,由于此蛋白的缺失,毛囊的分化不會(huì)被誘導(dǎo),休止期毛囊不會(huì)重新進(jìn)入生長(zhǎng)期,新的毛發(fā)也就無法再生長(zhǎng),進(jìn)而導(dǎo)致毛發(fā)的脫落。無毛蛋白功能缺失還可導(dǎo)致毛囊結(jié)構(gòu)的不成熟,從而引起多發(fā)性的擴(kuò)散性毛囊丘疹。
通過對(duì)文獻(xiàn)及數(shù)據(jù)庫(kù)的檢索,已經(jīng)發(fā)現(xiàn)了30余種不同的HR基因突變,包括不同的突變類型,如錯(cuò)義突變[5]、無義突變、缺失突變[6]和剪切位點(diǎn)突變[7,8]等。其中國(guó)外報(bào)道近親結(jié)婚的家系,APL的HR基因的突變均為純合突變。目前已發(fā)現(xiàn)的該基因的突變位點(diǎn)通常分散在整個(gè)HR基因上,還未發(fā)現(xiàn)熱點(diǎn)突變區(qū)域。本文報(bào)道的患兒家系否認(rèn)近親結(jié)婚史,患兒攜帶復(fù)合雜合突變,分別為移碼突變c.2270delC和錯(cuò)義突變c.3038T>C,在國(guó)內(nèi)外都屬首次報(bào)道。c.2270delC突變導(dǎo)致基因編碼框移碼,即p.S757Ffs*144,致使無毛蛋白截短;c.3038T>C引起患者無毛蛋白的第1013位的脯氨酸由亮氨酸替代,即p.P1013L,我們通過Polyphen-2軟件(http://genetics.bwh.harvard.edu/pph2/)對(duì)其進(jìn)行功能預(yù)測(cè),結(jié)果顯示該突變很可能是有損害的。這些突變可能導(dǎo)致無毛蛋白的功能異常,導(dǎo)致毛發(fā)周期不穩(wěn)定以及毛囊結(jié)構(gòu)不成熟,最終表現(xiàn)為毛發(fā)的脫失以及近膚色的毛囊丘疹。
我們應(yīng)用HR基因突變檢測(cè)在1例APL患兒中發(fā)現(xiàn)了2種新的HR基因突變c.2270delC和c.3038T>C,增加了中國(guó)漢族人群中HR基因的突變譜,為今后進(jìn)行產(chǎn)前診斷及遺傳咨詢奠定了基礎(chǔ)。
