基于結構導向集總的延遲焦化絕熱反應過程模型研究
2020-01-15 08:50:08倪騰亞劉紀昌
石油煉制與化工 2020年1期
關鍵詞:模型
葉 磊,汪 成,倪騰亞,劉紀昌,2,孫 輝,2
(1.華東理工大學化學工程聯合國家重點實驗室 上海 200237;2.綠色能源化工國際聯合研究中心)
延遲焦化作為最重要的原油二次加工工藝,由于原料組成和反應過程的復雜性,很難定量描述反應溫度、原料配比等操作條件對焦化產物分布的影響,因此工業應用中主要依靠操作經驗優化工藝,具有一定的盲目性。隨著原油重質化日益嚴重,僅憑經驗已不再適應發展要求,使得延遲焦化迫切需要精準模型來優化工藝。與此同時,計算機技術的不斷發展和分析測試方法的不斷改善為建立更精準模型創造了條件。目前,國內有不少學者結合計算機技術和分析條件針對延遲焦化工藝建立產物預測模型,例如范啟明等[1]建立的減壓重油延遲焦化反應6集總動力學模型、馬伯文等[2]建立的11集總反應動力學模型和盧山等[3]建立的12集總反應動力學模型等,但以上模型均屬于基于餾分的集總模型,預測精度較低,無法在分子水平上對延遲焦化過程進行產物分布預測和反應網絡計算。
結構導向集總方法(簡稱為SOL)由Jaffe等[4]于1992年首先提出,成功地將傳統的基于餾分的集總反應動力學模型在分子水平上應用和發展,并已經成功應用于加氫過程和催化裂化過程[5-6]。Jaffe和Ghosh等應用結構導向集總方法分別建立了預測汽油辛烷值和柴油十六烷值的模型,辛烷值和十六烷值的預測誤差均小于2%[7-8]。
孫忠超等[9]將SOL方法與Monte Carlo法相結合構建催化裂化汽油催化裂解動力學模型,該方法預測的產物分布誤差在10%以內。田立達等[10-11]建立了延遲焦化過程的結構導向集總反應動力學模型,可以用于延遲焦化裝置的產品產率和性質預測、原料優化配置等。
但上述基于結構導向集總方法的反應動力學模型皆采用等溫反應器模型,未考慮反應網絡的熱效應。實際生產中,延遲焦化在490~505 ℃的溫度區間內反應溫度每降低或升高1 ℃都會對反應產物分布產生明顯影響,而延遲焦化反應過程是以大分子生成小分子的吸熱反應為主,熱效應顯著。因此,將反應熱計入延遲焦化反應動力學模型顯得至關重要。
本課題在基于結構導向集總的分子尺度延遲焦化反應動力學模型基礎上,結合反應網絡中各反應速率和反應熱數據,建立延遲焦化絕熱反應動力學模型,通過對比模型預測結果與延遲焦化小型試驗產物產率和典型分子含量數據,驗證模型的可靠性。
1 延遲焦化原料分子組成矩陣的構建
針對典型延遲焦化體系油品的組成特點,考慮到其重金屬鎳、釩含量較高,基于結構導向集總方法,構建了包含鎳、釩在內的21個結構單元來描述延遲焦化原料分子組成。21個結構單元所代表的化學結構式如表1所示。
由結構單元可以構成任意一個延遲焦化體系油品分子的結構向量,每一個分子的結構向量包含21個元素,每個元素對應分子中包含的相應結構單元的數目。表2列舉了2個典型渣油分子(結構如圖1所示)的結構向量。
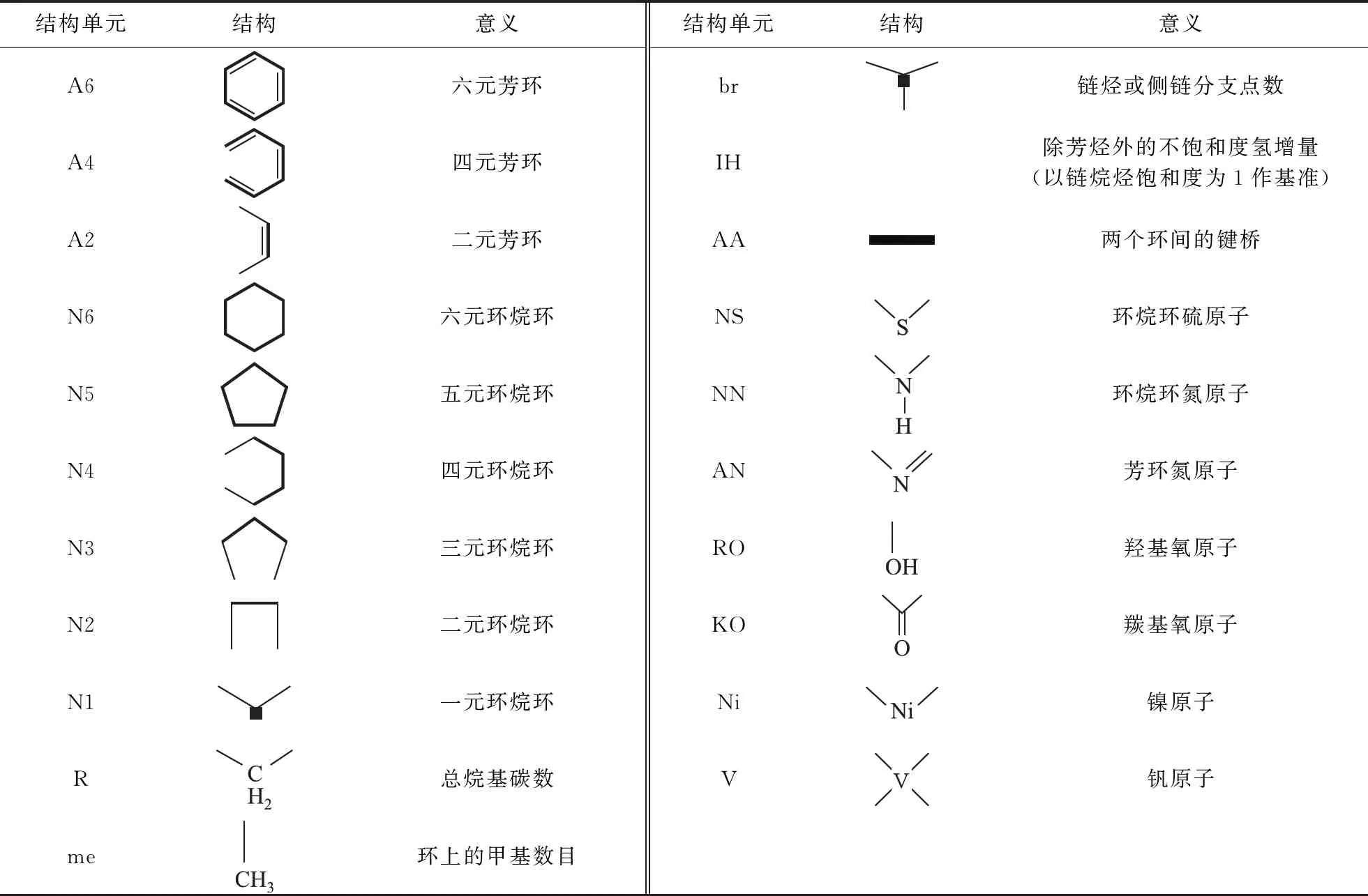
表1 21個結構單元的意義

圖1 2個典型渣油分子的結構式
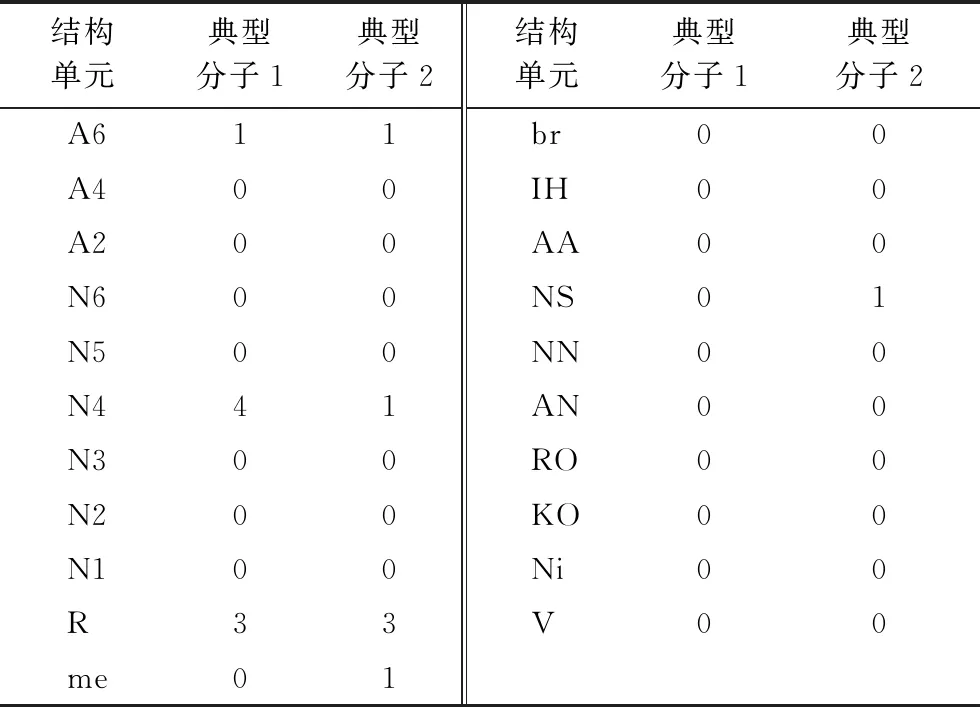
結構單元典型分子1典型分子2結構單元典型分子1典型分子2A611br00A400IH00A200AA00N600NS01N500NN00N441AN00N300RO00N200KO00N100Ni00R33V00me01
根據渣油原料分子組成,選取55類核心分子代表延遲焦化原料中的所有同系物[12],通過對核心分子添加0~50個—CH2—支鏈的方式,在刪除部分實際不存在的分子后,確定由2 791種分子的結構向量構成的2 791×21分子結構矩陣,并在矩陣最后添加一個代表對應分子摩爾分數、包含2 791個元素的列向量,形成一個2 791×22的延遲焦化原料分子組成矩陣。
為了建立延遲焦化原料的分子組成矩陣,首先采用紅外光譜、液相凝膠滲透色譜、氣相色譜-質譜、核磁共振等分析儀器對延遲焦化渣油原料的組成和結構進行精細表征,以得到詳盡的組成和性質數據(記作測試值),再采用基團貢獻法和渣油分子的結構向量理論計算得到相應的組成和性質數據(記作計算值)。然后,以測試值和計算值二者的殘差平方和構建目標函數,再結合模擬退火算法,利用Metropolis抽樣策略在解空間中進行搜索,最終得到一個近似全局最優解,由此得到一個組成和性質計算值與測試值重合度最高的分子組成矩陣。基于結構導向集總的渣油分子組成矩陣可由前期構建的模型計算獲得[13]。
2 延遲焦化過程結構導向集總模型的建立
2.1 延遲焦化過程的反應規則
結構導向集總中每個反應規則[14]都由以下兩部分構成:①反應物選擇規則,用來篩選出原料矩陣中能發生此類反應的分子;②產物生成規則,用來確定反應物分子發生反應后生成的產物分子。結合延遲焦化過程中的反應特征,制定了38條反應規則用于描述整個延遲焦化反應網絡,其中包括了脫氫、脫氫縮合、脫一氧化碳、脫二氧化碳、脫硫化氫、加氫脫氮、雙烯合成、碳鏈斷裂、側鏈斷裂和開環這10大類反應。
2.2 延遲焦化反應過程的反應網絡
將確立后的延遲焦化反應規則編譯成Matlab可識別的程序語言,并將渣油分子組成矩陣輸入至該程序中進行計算,根據反應物選擇規則判斷反應物可能發生的反應,并根據產物生成規則確定反應生成的產物,由此將渣油原料分子組成矩陣轉變為延遲焦化產物分子組成矩陣。計算后渣油原料分子組成矩陣中的每一個分子都形成了一個相應的從反應物到產物的反應網絡。由于從渣油原料分子為反應物到生成延遲焦化最終產物的反應網絡所包含的反應分子數目龐大、反應過程復雜,因此這里僅以甲基環己烷裂解反應為例簡要說明由反應網絡生成動力學微分方程組的過程。
圖2所示為甲基環己烷裂解的反應網絡。圖2中,甲基環己烷裂解反應網絡共涉及14種分子,每種分子用Yi(i=1~14)表示,其濃度用yi(i=1~14)表示。由于部分產物分子不包含在渣油分子組成矩陣中,所以通過在原料渣油分子組成矩陣的基礎上添加一些行向量,形成延遲焦化產物分子矩陣。甲基環己烷及其延遲焦化反應過程產物在熱裂化過程中可能發生13步反應,每步反應的速率常數用ki表示,第1、13步為脫氫反應,第2步為開環反應,第3步為側鏈全斷反應,第4、8步為脫氫芳構化反應,第5,6,7,9,10,11,12步為碳鏈斷裂反應。
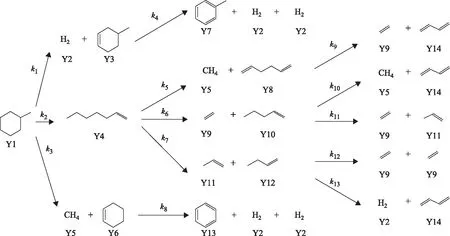
圖2 甲基環己烷裂解的反應網絡
基于延遲焦化自由基反應機理,假設所有反應為一級不可逆反應,共建立13個反應動力學方程,并將反應網絡中的所有分子表示為延遲焦化產物分子組成矩陣中其對應的行向量,得到如表3所示的甲基環己烷裂解反應網絡分子結構向量矩陣(矩陣中所有元素為零的列向量省略)和如表4所示的甲基環己烷裂解反應網絡反應物-產物對矩陣。
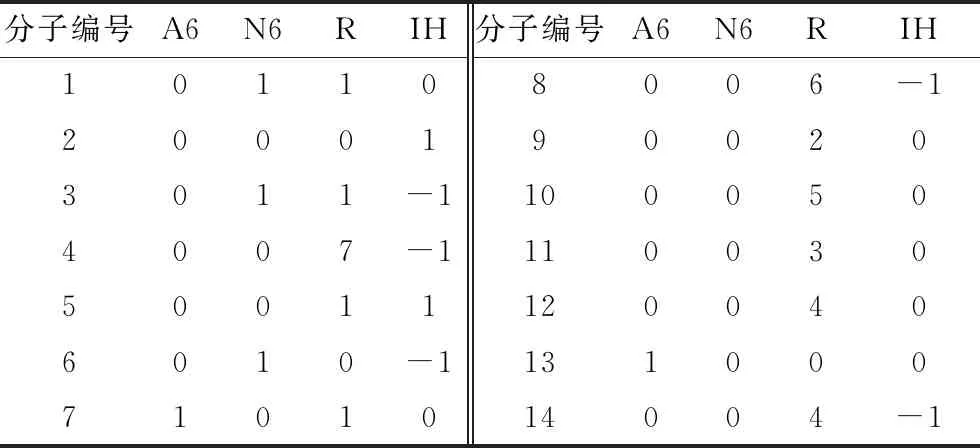
表3 甲基環己烷裂解反應網絡分子結構向量矩陣
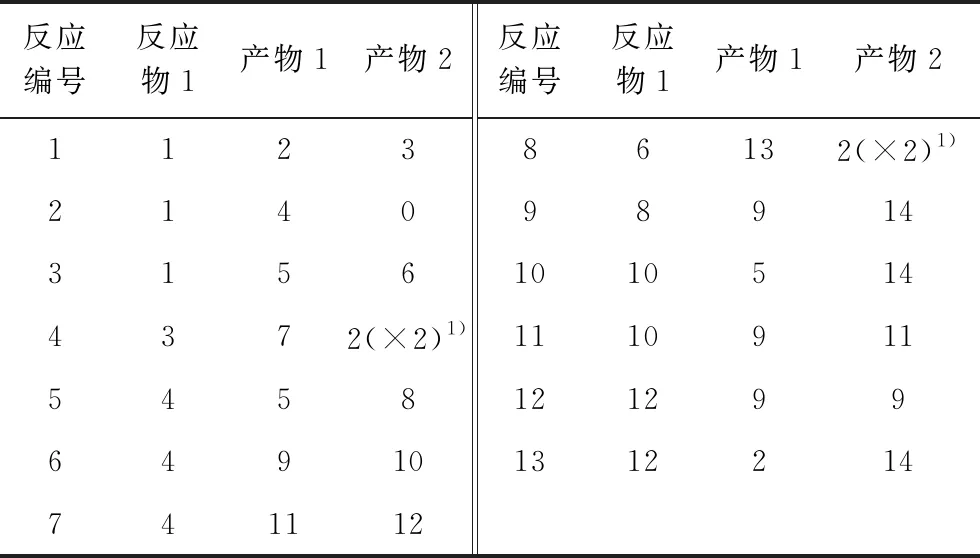
表4 甲基環己烷裂解反應網絡反應物-產物對矩陣
1)“×2”表示反應產物H2的分子數為2。
結合表4,由反應物-產物對可推出圖3所示的反應動力學方程式。

圖3 甲基環己烷反應網絡的反應動力學方程式
上述每種分子都涉及多個反應,即它們既可作一些反應的反應物,又可作一些反應的產物,在合并各分子的反應方程式后,得到如圖4矩陣形式所示的甲基環己烷反應網絡的反應動力學微分方程組。
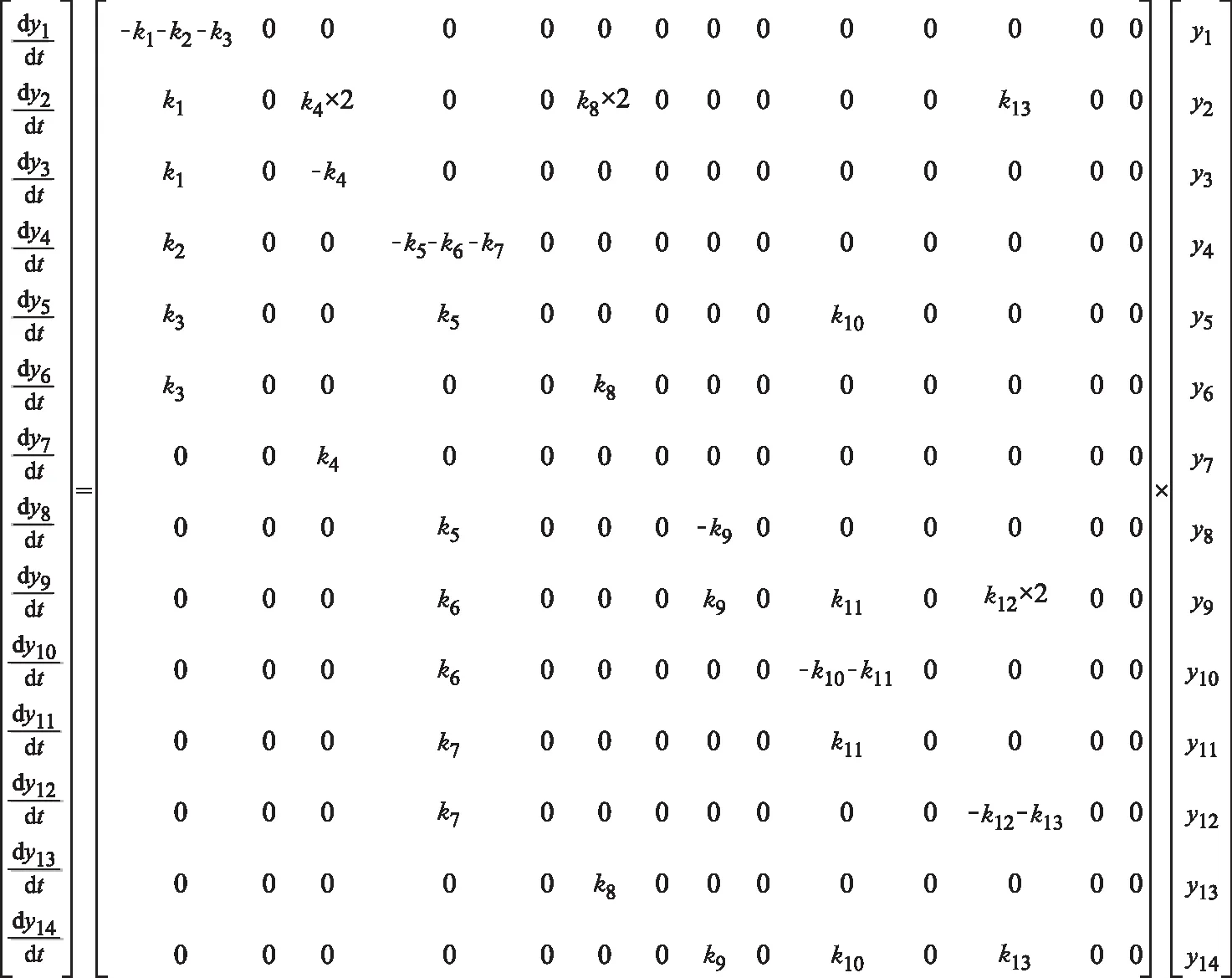
圖4 甲基環己烷反應網絡動力學微分方程組的矩陣形式
2.3 延遲焦化反應過程的熱效應
2.3.1 延遲焦化過程的絕熱反應器模型焦炭塔是延遲焦化工藝最重要的設備,渣油等延遲焦化原料在焦炭塔中轉化成價值更高的汽油、柴油和蠟油等輕質油品。原料經加熱爐升溫至一定溫度,進入焦炭塔后主要發生各種大分子裂解成小分子的反應,沒有外界能量的輸入,屬于絕熱反應過程,因此焦炭塔內的溫度沿物流方向呈降低趨勢。
建立延遲焦化絕熱反應模型時,為了方便計算,制定3條合理假設:①反應由多個微元反應時間段構成,在每個微元段內反應溫度恒定不變,后續微元段的反應溫度的變化值由上一微元段的反應熱計算得到;②反應原料由塔底勻速上升,液面上部原料裂解氣體不與焦炭塔內壁進行熱量交換,液面下部原料液體不與焦炭塔內壁進行熱量交換;③焦炭塔內的軸向和徑向的熱量傳遞不計入模型計算中。
2.3.2 熱裂化反應熱效應計算反應熱代表的是化學反應過程中吸收或釋放的能量。在恒壓條件下,反應熱等于反應體系的焓變。當反應物的能量之和大于生成物的能量之和時,反應為放熱反應;若反應物的能量之和小于生成物的能量之和,則反應為吸熱反應。由于模型計算涉及的反應數目巨大,絕大部分無法通過查閱文獻獲得反應熱數據,因此需借助于量子化學計算軟件獲得反應網絡中所有反應的反應熱。本課題采用Materials Studio(MS)軟件進行計算,通過構建分子結構,在進行幾何構型優化的基礎上,獲得反應物分子和產物分子能量最低的空間構型,從而得到反應物分子和產物分子的最低能量。
以正丁基苯裂解生成乙苯和乙烯為例,正丁基苯分子結構進行幾何優化后能量為-13.1 kJmol,乙苯和乙烯分子結構進行幾何構型優化后,能量之和為82.4 kJmol,生成物和反應物能量之差ΔH為95.5 kJmol,與文獻值104.4 kJmol相吻合[15]。表5列出了一些烴類在溫度為500 ℃下裂解的反應熱計算值和文獻值的對比。

表5 500 ℃下部分烴類裂解的反應熱文獻值和計算值的對比
實際模型計算中,反應數量龐大,很難對每個反應進行準確的反應熱計算。因此,用式(1)擬合生成物和反應物的能量之差ΔH。
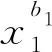
(1)
式中:ai和bi為回歸待定參數;xi為某一分子結構單元;ΔH0為常量。
利用MS軟件的Dmol3模塊進行計算,得到反應的ΔH數值。以此為據進行數據回歸計算,根據不同焦化反應類型選取具有代表性的結構單元作為xi,利用軟件Matlab中最優化工具箱的lsqcurvefit函數,回歸待定參數ai和bi,由此計算得到不同類型反應的ΔH關聯式。部分反應的反應熱關聯式如下。
①碳鏈斷裂:
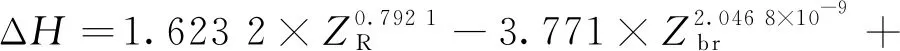
②脫氫:
③雙烯合成:
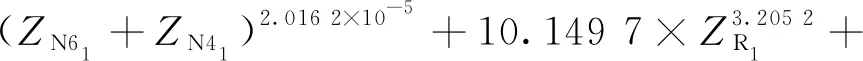
式中,Z表示油品分子中包含的相應結構單元的數目,下標表示結構單元,下同。
2.4 延遲焦化結構導向集總模型的求解
2.4.1 反應速率常數的求取本課題進行延遲焦化反應網絡計算時采用文獻[16]的熱裂化反應速率常數,參照反應ΔH的處理方法,不同反應類型反應速率常數關聯式為:
(2)
式中:ΔE為反應物和過渡態之間的能壘,kJmol;ΔSm為反應物和過渡態之間的熵變,kJ(mol·K);kB為玻爾茲曼常數;h為普朗克常數;R為氣體狀態常數;T為溫度,K。由式(2)求解速率常數k(T),必須先求解ΔE和ΔSm,以下為碳鏈斷裂、脫氫和雙烯合成反應的ΔE和ΔSm求解公式。
①碳鏈斷裂
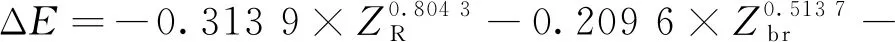

②脫氫

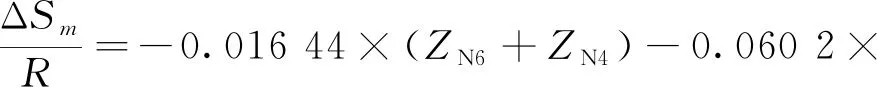
③雙烯合成
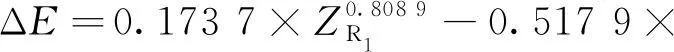
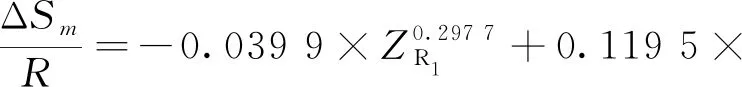
2.4.2 反應網絡的求解基于上述研究,將整個焦化反應時間t劃分成n個微元反應時間段Δti(i=1~n)。在第n個微元反應時間Δtn內,輸入原料分子組成矩陣與焦化反應溫度T,基于結構導向集總方法的反應規則建立反應網絡,根據關聯式計算得到的反應速率常數,構建反應動力學微分方程組,利用改進的Runge-Kutta法計算得到產物分子組成矩陣。將產物分子組成矩陣與未反應的原料分子組成矩陣合并后,得到第n+1段微元反應時間段Δtn+1的原料分子組成矩陣,并計算微元時間Δtn內所有反應的反應熱之和,換算成溫差ΔTn,得到第n+1段微元反應時間Δtn+1的焦化反應溫度T+ΔTn,進行Δtn+1內的焦化反應過程計算。依次循環計算,直到焦化反應時間為t。
3 延遲焦化工藝SOL絕熱反應過程模型的驗證
延遲焦化小型試驗在華東理工大學石油加工研究所延遲焦化及產物精餾小型試驗裝置上進行。裝置的焦化塔體積為4 L,精餾釜體積為2 L。試驗條件為:渣油原料進料量1 000 gh,去離子水進料量20 gh,共同進料時間3 h,渣油進料結束后,繼續進去離子水汽提2 h,焦炭塔中反應溫度470~505 ℃,反應壓力0.15 MPa,循環比(指循環蠟油質量流量與原料油質量流量的比值)0~0.6。
試驗結束后,基于結構導向集總反應動力學的延遲焦化絕熱反應器模型,通過輸入原料渣油分子矩陣與延遲焦化工藝條件,計算后可獲得延遲焦化過程中分子組成變化和產物分布。通過將模型預測值與延遲焦化實驗室小型試驗數據進行對照,以驗證模型的可靠性。
3.1 延遲焦化SOL絕熱反應器模型與等溫反應器模型的對比
延遲焦化原料經加熱爐加熱至設定溫度后,進入焦炭塔發生熱裂化反應,塔內大量分子的裂解反應熱將引起反應體系溫度的明顯變化。將渣油的平均摩爾熱容引入延遲焦化反應器模型,計算得到每一反應的反應熱,從而獲得整個延遲焦化體系反應前后的溫度變化。在不同反應條件下,不同渣油反應前后反應體系的溫度變化如表6所示。
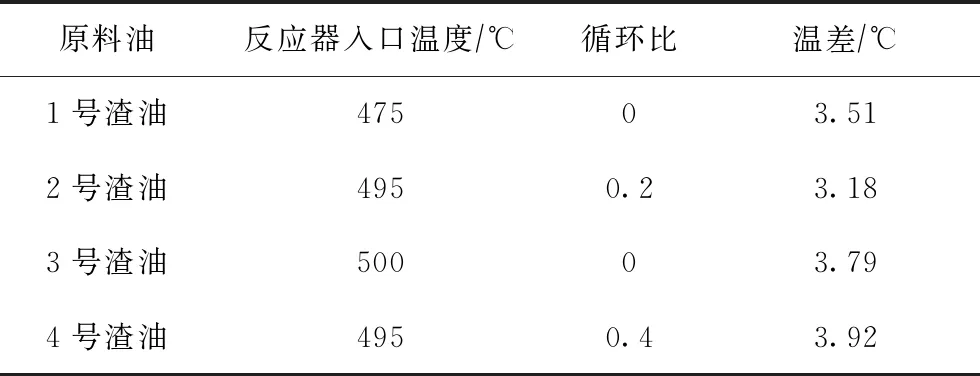
表6 不同渣油原料反應前后體系溫度的變化
由表6可知,渣油延遲焦化反應前后溫差達3~4 ℃,焦化反應前后體系溫度相差明顯,因此將反應熱效應計入延遲焦化模型是十分必要的。本研究考慮到反應熱效應引起的溫度變化,建立絕熱反應器模型,對1號渣油和2號渣油的延遲焦化反應過程進行模擬計算,并與等溫反應器模型的模擬結果及實驗室小型試驗結果進行對比,產物分布的對比如表7所示。其中,1號渣油延遲焦化反應條件為:入口溫度495 ℃、循環比0;2號渣油延遲焦化反應條件為:入口溫度495 ℃、循環比0.4。
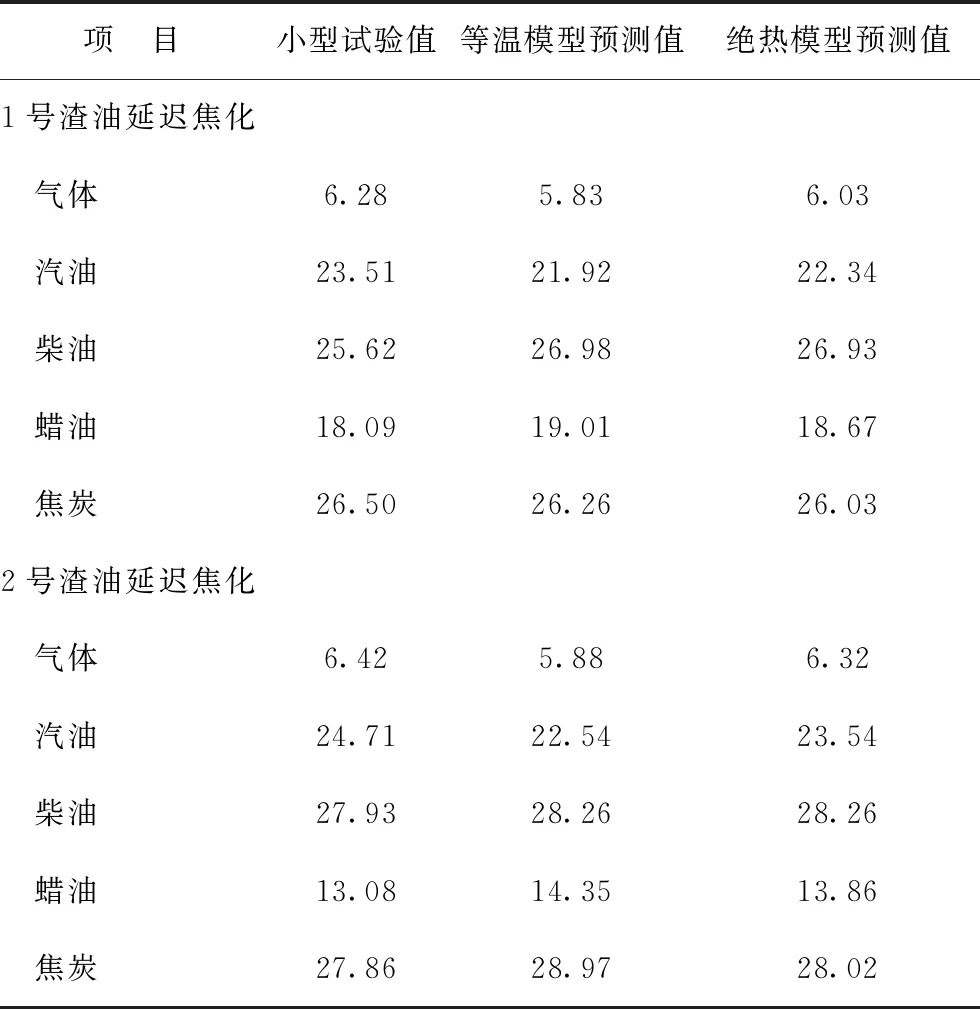
表7 絕熱、等溫反應器模型計算的產物分布與實驗室小型試驗結果的對比 w,%
由表7可知,將實驗室延遲焦化小型試驗數據作為基準,由絕熱反應器模型計算獲得的延遲焦化產物中的氣體、汽油、柴油、蠟油和焦炭5種產物的產率比未計入反應熱效應的等溫反應器模型的預測誤差更小。采用結構導向集總反應動力學模型,通過計算反應網絡中典型反應的熱效應來描述焦化塔中的溫度變化,并將反應速率與反應熱效應耦合,使模型預測實際延遲焦化過程的反應規律更加準確。
采用絕熱、等溫反應器模型計算獲得的典型分子含量與實驗室小型試驗結果的對比如表8所示。由表8可知,將實驗室延遲焦化小型試驗數據作為基準,由絕熱反應器模型計算獲得的延遲焦化產物中甲烷、乙烷、丙烷、氫氣、正庚烷等典型分子的含量比由等溫反應器模型計算的結果誤差更小,進一步證明延遲焦化絕熱反應器模型的預測精度優于延遲焦化等溫反應器模型。
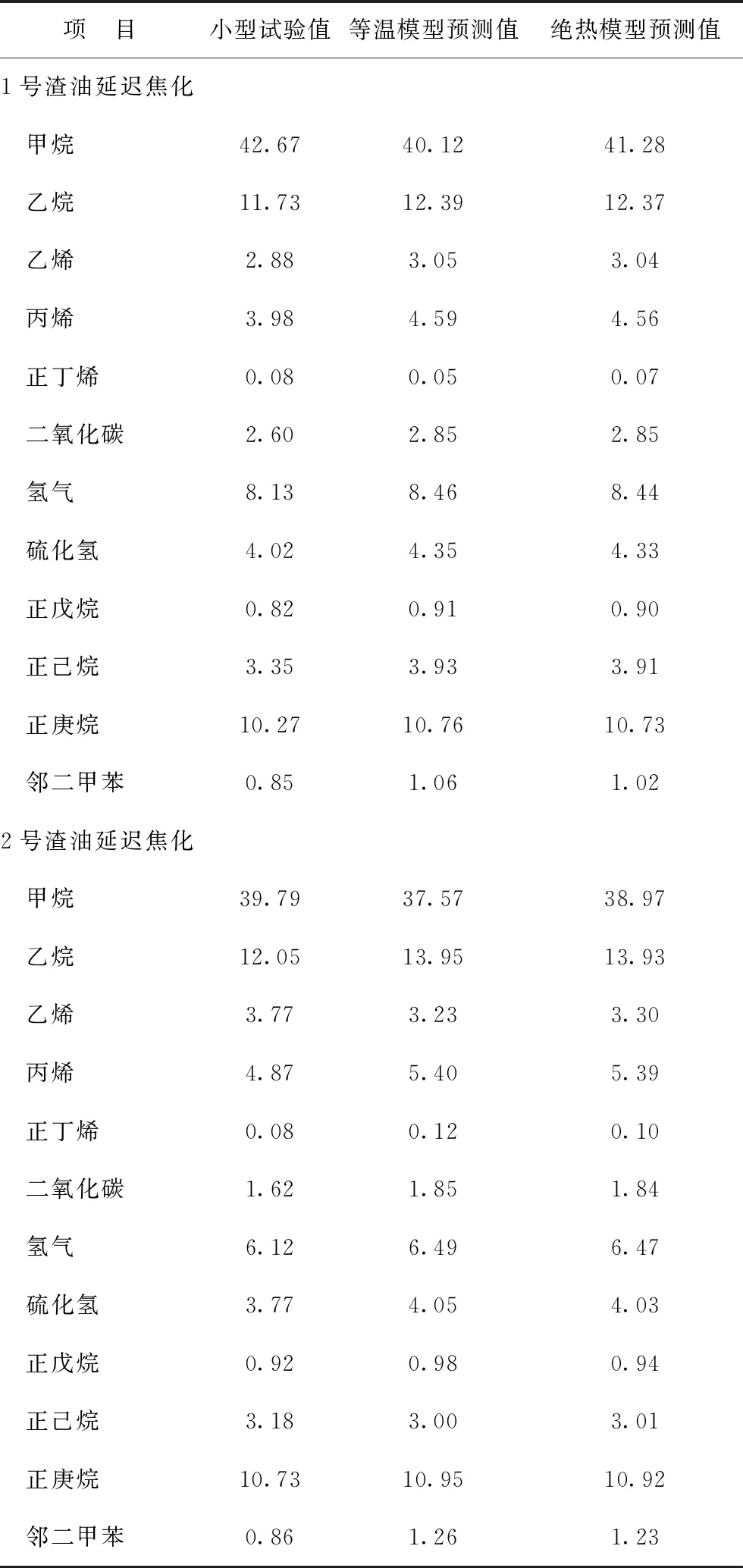
表8 絕熱、等溫反應器模型計算的典型分子含量與實驗室小型試驗結果的對比 w,%
3.2 延遲焦化SOL絕熱反應器模型對不同溫度下焦化反應的預測
3.2.1 延遲焦化SOL絕熱反應器模型預測不同反應溫度下的產物分布在循環比為0、反應溫度為470~505 ℃(間隔為5 ℃)的反應條件下,對1號渣油原料進行延遲焦化小型試驗,并通過延遲焦化SOL絕熱反應器模型進行模擬計算,所得氣體、汽油、柴油、蠟油和焦炭5種產物產率的模型預測值與試驗值的對比如圖5所示。
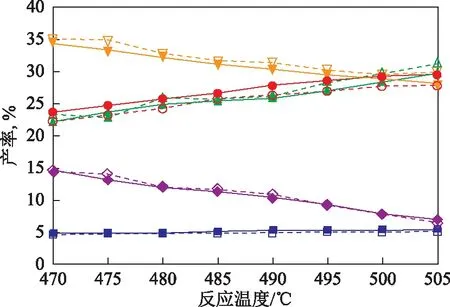
圖5 1號渣油原料在不同反應溫度下延遲焦化的產物產率模型預測值與試驗值的對比■—氣體產率預測值; ●—汽油產率預測值; ▲—柴油產率預測值; 蠟油產率預測值; ◆—焦炭產率預測值; □—氣體產率試驗值; ○—汽油產率試驗值; △—柴油產率試驗值; 蠟油產率試驗值; ◇—焦炭產率試驗值
由圖5可以看出,通過延遲焦化SOL絕熱反應器模型預測得到的氣體、汽油、柴油、蠟油和焦炭5種產物的產率與小型試驗數據較為接近,預測誤差均未超過1.7百分點。表明該模型可用于預測渣油在不同反應溫度下延遲焦化所得氣體、汽油、柴油、蠟油和焦炭的產率。
3.2.2 延遲焦化SOL絕熱反應器模型預測不同反應溫度下焦化產物中典型分子含量使用色譜分析儀器分析不同反應溫度下1號渣油延遲焦化反應所得氣體和汽油中的典型分子含量,并與SOL絕熱反應器模型預測值進行對比,結果如表9所示。
由表9可以看出,在不同反應溫度條件下,1號渣油原料油的焦化產物中典型分子含量的SOL絕熱反應器模型預測誤差均在2.0百分點以內,與小型試驗數據較為接近,這表明延遲焦化SOL絕熱反應器模型可用于預測在不同反應溫度條件下焦化產物中典型分子的含量。
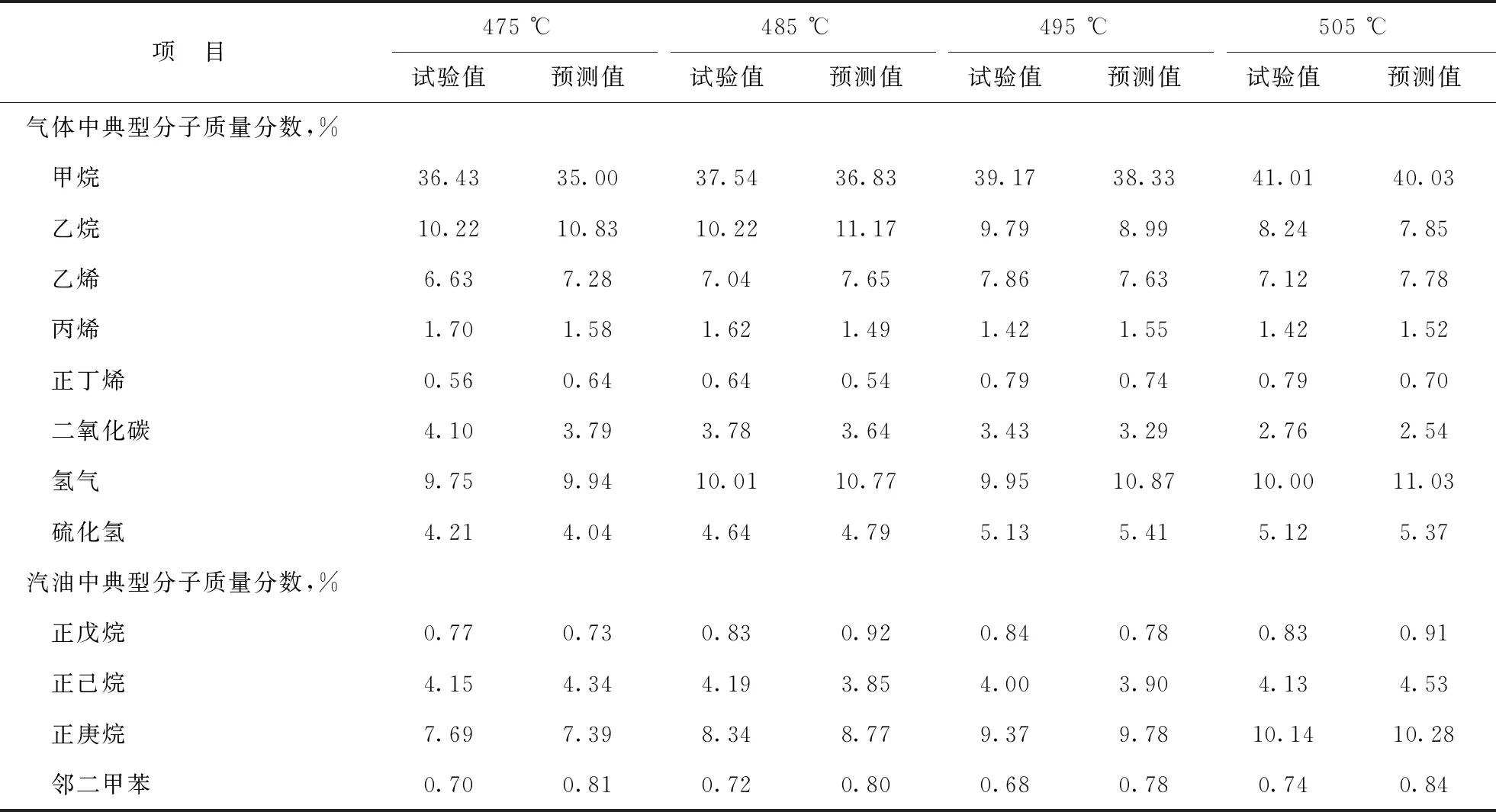
表9 1號渣油原料不同反應溫度下焦化產物中典型分子含量的模型預測值與試驗值的對比
4 結 論
(1)基于結構導向集總方法和延遲焦化反應機理,制定了包含脫氫、脫氫縮合、脫一氧化碳、脫二氧化碳、脫硫化氫、加氫脫氮、雙烯合成、碳鏈斷裂、側鏈斷裂和開環等10類38條反應規則,用于描述延遲焦化過程的反應網絡。結合相應的反應速率常數和反應熱數據,建立了反應動力學微分方程組,通過改進的Runge-Kutta法求解,構建出基于結構導向集總的延遲焦化絕熱反應動力學模型,預測延遲焦化過程的典型分子組成和產物分布。
(2)基于結構導向集總動力學對延遲焦化絕熱反應器模型進行計算,結果表明渣油延遲焦化反應前后溫差達3~4 ℃,焦化反應結束溫度與進料溫度差異明顯,因此考慮反應熱效應對延遲焦化過程的影響是完全有必要的。延遲焦化SOL絕熱反應器模型的預測精度優于未計入反應熱效應的等溫反應器模型。
(3)采用延遲焦化SOL絕熱反應器模型對1號渣油原料油在反應溫度470~505 ℃下的延遲焦化反應過程進行模擬,計算得到的延遲焦化氣體、汽油、柴油、蠟油和焦炭5種產物的產率與延遲焦化小型試驗數據較為接近,預測誤差不超過1.7百分點,焦化產物中典型分子含量與延遲焦化小型試驗數據較為接近,預測誤差在2.0百分點以內。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
