HPLC-PAD法優化烏骨雞活性肽分離純化
2019-12-30 05:01:24李映紅吳正治黃飛娟
生物加工過程 2019年6期
關鍵詞:優化
白 芳,李映紅,吳正治,黃飛娟,3
(1.深圳大學附屬第一醫院,廣東 深圳 518035;2.深圳市老年醫學研究所,廣東 深圳 518020;3.中山大學附屬第一醫院,廣東 廣州 510080)
烏骨雞(Black-Bone Silky Fowl,GallusgallusdomesticusBrisson),是一種具有藥用價值的雞種。研究證明,烏骨雞對各類虛癥,如氣虛、血虛、脾虛、腎虛和心虛等均有良好療效[1-3]。烏骨雞活性肽,即烏骨雞肌肉蛋白通過一定優化工藝酶促水解后獲得的具有較小分子量范圍的多肽產物,分子量集中在3 000以下,其營養價值高、更易吸收[4-5]。烏骨雞多肽內部普遍存在功能區,這些功能區的研究對于烏骨雞活性肽臨床價值的充分利用尤其重要。然而,烏骨雞活性肽的分離純化是研究其應用價值的基礎。
目前用于烏骨雞多肽的分離技術最為常見的有凝膠過濾色譜法和反相液相色譜法,常用的分離體系為甲醇和乙腈體系等[6-9]。根據多肽混合物進入分析級反相色譜柱時表現的極性和進入凝膠色譜柱時分子量的不同進行高效液相色譜(HPLC)的分離純化[10]。反相色譜柱分離純化效果一般較凝膠色譜柱好,但不能表現出多肽分子量通過色譜柱的前后次序;乙腈比甲醇分離效果好,但其價格昂貴[11]。在多肽的分離純化中,一般將凝膠過濾色譜法和反相液相色譜法結合使用[12-13]。然而,根據這兩種不同原理,選擇不同流動相及其他參數等的各自分離條件及分離效果如何,并沒有統一性。
為解決上述多肽分離純化的共性問題,同時提供藥用價值突出的烏骨雞多肽的最佳分離條件,本研究中,筆者主要針對目前常用酶解工藝流程制備烏骨雞活性肽,即木瓜蛋白酶和風味蛋白酶對烏骨雞肌肉蛋白進行充分酶解后,采用高效液相色譜(配置PDA全波長檢測器)(HPLC-PDA)法對C18反相色譜柱(0.1%三氟乙酸或磷酸鈉/甲醇/水體系)及TSKgel凝膠色譜柱(0.1%三氟乙酸/乙腈/水體系)的多肽分離條件分別進行優化,從流動相、物料比、流速、進樣量、波長等方面進行探究,以出峰數為主,結合分離度、峰形、基線、信號等考察其分離效果。
1 材料與方法
1.1 主要試劑
食品級木瓜蛋白酶和風味蛋白酶,廣州裕立寶科技有限公司;BCA蛋白檢測分析試劑盒,美國Thermo公司;三氟乙酸、甲醇及乙腈為色譜純,美國Sigma公司;NaOH及Na3PO4為國產分析純,廣東光華科技股份有限公司。
1.2 主要儀器
膠體磨,南通澳德精密機械公司;酶標儀,美國Thermo公司;島津LC20 AD型高效液相色譜儀(配置PDA全波長檢測器)、C18色譜柱Inertsil SOD-SP(5 μm,150 mm×4.6 mm)及凝膠色譜柱TSKgel G2000 SWxl (5 μm,300 mm×7.8 mm),日本島津公司。
1.3 烏骨雞活性肽制備
選取江西省泰和烏骨雞,體質量1.5 kg左右,雌雄各半(各10只)。復合酶解法制備烏骨雞活性肽[14]。烏骨雞肌肉蒸煮后,絞碎再加入膠體磨進一步磨碎,加入木瓜蛋白酶(4 000 U/g)和風味蛋白酶(2 000 U/g)進行酶解,其中酶解溫度為55 ℃,酶解時間為3.5 h,沸水高溫保持20 min進行滅酶,4 000 r/min離心15 min,分去上層油脂和下層沉淀物,取中層液,即為烏骨雞活性肽溶液。按照BCA蛋白檢測試劑盒的操作說明書進行蛋白定量,結果為1.597 mg/mL。
1.4 反相液相色譜法優化烏骨雞活性肽分離條件
選擇C18反相液相色譜法常用分離體系[15]:0.1%三氟乙酸或pH 6.0磷酸鹽/甲醇/水體系,對烏骨雞活性肽進行流動相物質配比、流速、進樣量及波長的選擇。首先,設置流動相體系為0.1%三氟乙酸/甲醇/水體系,甲醇體積分數設為20%、10%、8%、6%,對應流速依次為0.8、0.5、0.3和0.2 mL/min,同時選擇進樣量10 μL、柱溫30 ℃、進樣室溫度4 ℃、測試時間30 min進行紫外全波長掃描。然后,流動相0.1%三氟乙酸/6%甲醇/水體系中更換三氟乙酸為磷酸鹽緩沖液,調節磷酸鹽緩沖液pH 至 6.0,進行紫外全波長掃描。此時,進樣量分別設為5、10、50 μL,流速為0.2 mL/min,其他條件如上。
1.5 凝膠過濾色譜法優化烏骨雞活性肽分離條件
TSKgel凝膠過濾色譜法進行烏骨雞活性肽的分離[16],首先選擇甲醇/水體系進行分離優化,但優化效果不理想,更換為0.1%三氟乙酸/乙腈/水體系進行分離優化。采用0.1%三氟乙酸/15%乙腈/水均勻無梯度洗脫,進樣量設為10 μL、流速0.5 mL/min、柱溫30 ℃、進樣室溫度4 ℃、測試時間30 min進行紫外全波長掃描。然后,采用0.1%三氟乙酸/(15%~65%)或(10%~25%)乙腈/水體系均勻上升梯度洗脫30 min,同時設置不同流速或不同進樣量進行優化,進行紫外全波長掃描。
1.6 數據處理
采用BCA蛋白檢測試劑盒重復檢測3次,外標法進行樣品蛋白的定量。
2 結果與討論
2.1 反相液相色譜法分離烏骨雞活性肽的優化結果分析
2.1.1 反相液相色譜法最佳優化結果
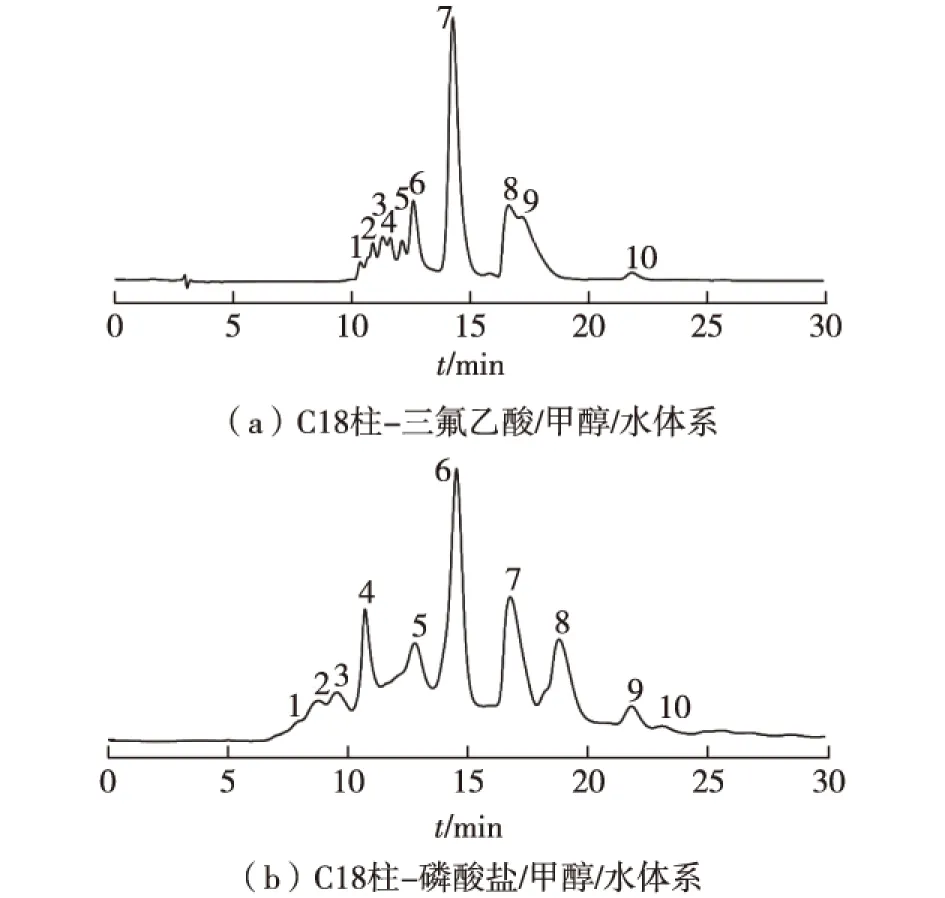
圖1 C18反相液相色譜法優化烏骨雞活性肽的分離條件Fig.1 Optimizing isolation condition of peptides by C18 reversed-phase chromatography
三氟乙酸及磷酸鹽作為緩沖液時,反相液相色譜法分離烏骨雞活性肽的最佳分離情況如圖1所示。由圖1可知:在分離小分子多肽時,反相液相色譜法的實驗優化結果為C18柱-0.1%三氟乙酸或pH 6.0磷酸鹽/6%甲醇/水體系-流速0.2 mL/min-進樣量5或10 μL-波長254 nm(10 μL)或220(5 μL)。此處,若結合分離度進行選擇時,應選擇C18柱-pH 6.0磷酸鹽/6%甲醇/水體系-流速0.2 mL/min -進樣量5 μL -波長220 nm,此時出峰距離相對較大,分離度和峰形均較好。
2.1.2 反相液相色譜法優化過程分析
本實驗中,筆者主要考察C18色譜柱、緩沖液、甲醇、流速、進樣量和波長等指標對烏骨雞多肽分離效果的影響。在常用流動相0.1%三氟乙酸/甲醇/水體系中固定其他條件,變換甲醇濃度和流速大小,優化過程如表1所示。由表1可知:隨著甲醇含量降低,流速降低,出峰數增加;當甲醇體積分數6%、流速0.2 mL/min時,分離效果最好。當把三氟乙酸換為磷酸鹽(6%甲醇/水中加入4% 0.02 mol/L NaH2(PO4)3溶液,H3PO4調節pH至6.0)時,流速0.2 mL/min,當三氟乙酸或磷酸鹽作為緩沖液時,出峰數基本一致,但三氟乙酸作為緩沖液時分離峰相對集中。進樣量為50 μL時,220 nm處吸收較強,254 nm處分離效果較好;進樣量為5 μL時,220 nm處分離效果較好,254 nm處吸收弱。190 nm處均無吸收,在280 nm處總體出峰數不如254 nm處的效果。
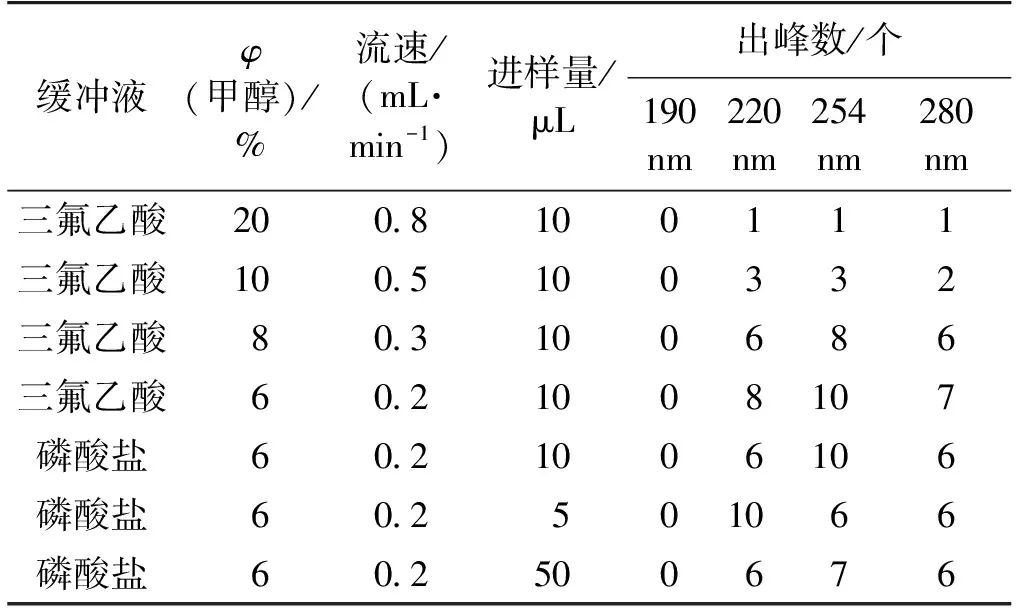
表1 反相液相色譜法分離烏骨雞活性肽的優化過程
2.1.3 反相液相色譜法優化烏骨雞活性肽分離條件的應用
通常對一次分離中分辨率不好的親水性多肽混合物進行二次分離,可以得到多個新肽,同時放大到制備級HPLC中,結合質譜共同引導實現多肽的全自動化分離和收集[13]。由于C18反相色譜柱的柱效高,TSKgel凝膠色譜柱可以用來確定分離物分子量分布,實際實驗中通常將優化的分析級反相色譜法和凝膠色譜法相互結合使用[17]。本實驗中,C18反相液相色譜法采用甲醇/水體系進行優化分析,需要更為精細的優化時,可將其梯度洗脫體系或乙腈體系作為流動相代替其進行進一步分離純化。
2.2 凝膠色譜柱過濾色譜法分離烏骨雞活性肽的優化結果分析
2.2.1 凝膠過濾色譜法優化結果
TSKgel-三氟乙酸/乙腈/水體系中,研究不同波長條件下TSKgel凝膠過濾色譜法優化烏骨雞活性肽,結果如圖2所示。由圖2可知:在分離小分子多肽時,凝膠過濾色譜法的實驗優化結果為TSKgel柱-0.1%三氟乙酸/(10%~25%)乙腈/水體系-流速0.5 min/mL-進樣量5 μL-波長220 nm。若不考察出峰數,只考察分離度和峰形進行選擇時,應選擇的優化結果為TSKgel柱-0.1%三氟乙酸/(15%~65%)乙腈/水體系-0.5 min/mL流速-進樣量10 μL -波長254 nm,此時出峰數較少,但分離度和峰形都較好。
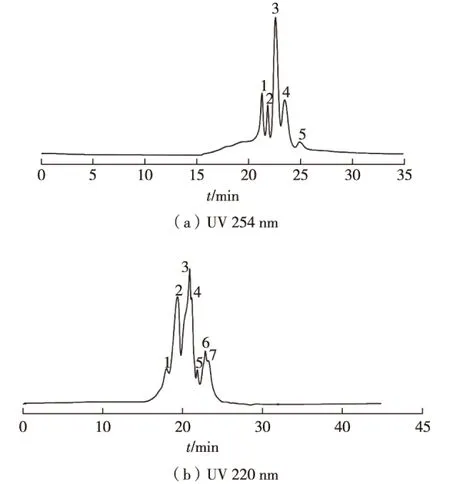
圖2 TSKgel凝膠過濾色譜法優化烏骨雞活性肽的分離條件Fig.2 Optimizing isolation condition of peptides by TSKgel filtration chromatography
2.2.2 凝膠過濾色譜法優化過程分析
主要考察凝膠色譜柱、乙腈、流速、進樣量和波長等指標對烏骨雞多肽分離效果的影響,優化過程如表2所示。由表2可知:乙腈梯度洗脫時,分離效果最好,隨著梯度中乙腈含量降低至一定程度,出峰數增加至最大。當變換流速時,流速為0.4或0.5 mL/min時,分離效果一致好于流速為0.3 mL/min時。當進樣量為5、10 μL時,波長對峰形、出峰數幾乎無影響。波長為190 nm時樣品均無吸收,且當波長為254和280 nm時,樣品的吸收強度較小。
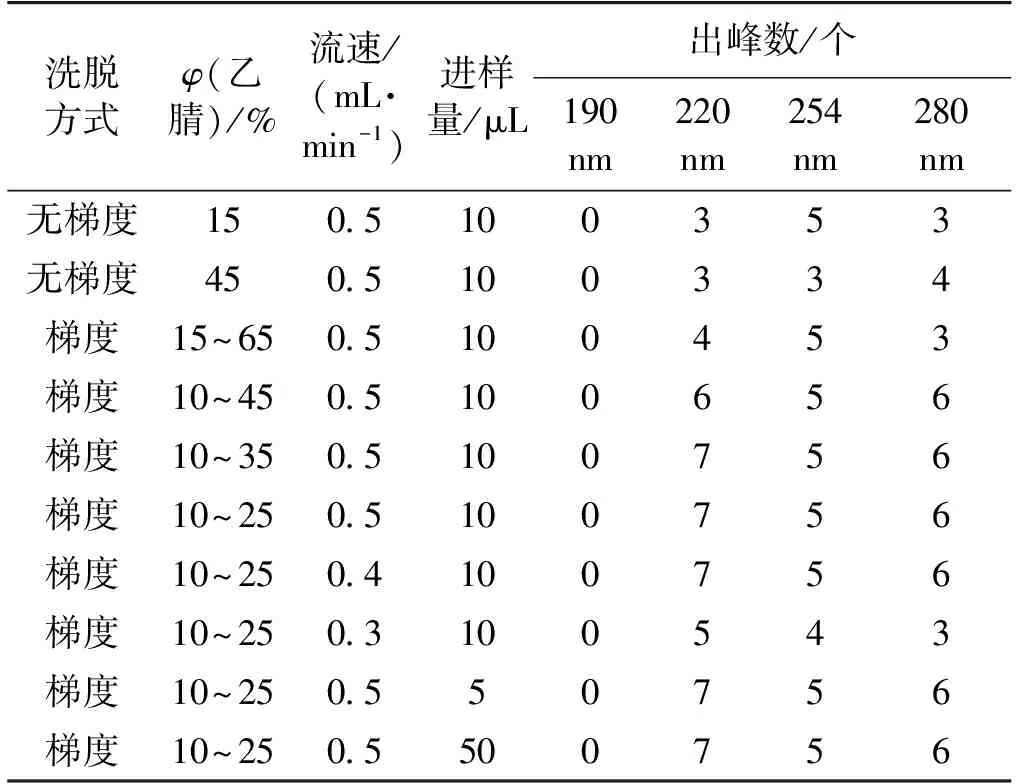
表2 凝膠過濾色譜法分離烏骨雞活性肽的優化過程
2.2.3 凝膠過濾色譜法優化烏骨雞活性肽分離條件的應用
在使用凝膠色譜柱-三氟乙酸/乙腈/水體系之前,筆者采用了價格低廉的三氟乙酸/甲醇/水體系的梯度洗脫條件分離烏骨雞活性肽,但該體系分離烏骨雞多肽效果不理想,可能是由于凝膠柱的柱效差或甲醇洗脫效果不好等因素導致[18],故采用乙腈/水體系進行分離條件優化。另外,在采用凝膠色譜法分離時,并未使用磷酸鹽作緩沖液,是因為三氟乙酸作緩沖液時,未見相對集中的分離峰出現。根據峰的保留時間分段收集凝膠色譜法分離樣品后得到的餾分,再結合C18反相色譜法進行餾分分離,這樣可實現多肽不同分子量段的深入分離、純化及分析等[17]。當混合多肽過于復雜、目標物質相對豐度較低時,可先使用本實驗已經優化的凝膠色譜法的分離條件,然后采用甲醇或乙腈梯度洗脫體系繼續優化C18色譜分離條件,從而實現目標物質的純化。
3 結論
反相液相色譜法分離小分子多肽的優化條件:C18柱-pH 6.0磷酸鹽/6%甲醇/水體系-流速0.2 mL/min-進樣量5 μL-波長220 nm。凝膠過濾色譜法分離小分子多肽的優化條件:TSKgel柱-0.1%三氟乙酸/(10%~25%)乙腈/水體系-流速0.5 mL/min-進樣量5 μL-波長220 nm。當使用反相液相色譜法分離小分子多肽時,隨甲醇含量及流速的降低,出峰數增加;三氟乙酸或磷酸鹽作緩沖液時出峰數幾乎一致,但分離度不同。凝膠色譜法分離小分子多肽時,乙腈梯度洗脫較無梯度洗脫分離效果好,且乙腈梯度濃度降低到一定程度后,分離效果不變。該研究為烏骨雞多肽及多肽類天然食品的精細分離純化提供參考。
猜你喜歡
房地產導刊(2022年5期)2022-06-01 06:20:14
能源工程(2022年1期)2022-03-29 01:06:28
建材發展導向(2021年12期)2021-07-22 08:06:48
建材發展導向(2021年7期)2021-07-16 07:07:52
中學生數理化(高中版.高二數學)(2021年12期)2021-04-26 07:43:48
中學生數理化(高中版.高考數學)(2021年12期)2021-03-08 01:28:50
今日農業(2020年16期)2020-12-14 15:04:59
消費導刊(2018年8期)2018-05-25 13:20:08
家庭影院技術(2018年4期)2018-05-09 07:07:41
電子制作(2017年20期)2017-04-26 06:57:45
