GC-MS/MS法測定煙草中的57種酯類香味成分
2019-12-28 03:30:32趙嘉幸任宗燦王曉瑜崔華鵬謝復煒劉惠民
煙草科技 2019年12期
趙嘉幸,陳 黎,任宗燦,王曉瑜,崔華鵬,謝復煒,劉惠民
中國煙草總公司鄭州煙草研究院,鄭州高新技術產業開發區楓楊街2 號 450001
酯類香味物質是煙草中常用的一類香料,對卷煙風味有重要貢獻。研究表明,單體酯類通常具有甜味、果香或葡萄酒香氣,與煙草香氣協調;混合酯類可賦予煙氣特定的氣味,抑制刺激性,使煙草香味柔和,并且在適宜用量范圍內一般不會對卷煙香氣有負面作用[1-3]。然而,目前對煙草中重要酯類香味成分的檢測方法鮮見報道。煙草組成成分繁雜,且大部分酯類香味成分質量分數低,因此高效靈敏的檢測手段必不可少。
目前對煙草中香味成分的提取方法主要包括水蒸氣蒸餾法[4]、固相微萃取法[5]、吹掃捕集法[6]、超臨界萃取法[7]、頂空萃取法[8]、同時蒸餾萃取法[9]等,這些方法存在加熱時間長或費用昂貴等問題。QuEChERS 前處理方法具有快速、簡便、廉價、高效、安全、低溫等優點[10],目前已廣泛用于農藥或獸藥殘留、法醫鑒定、毒品檢測以及環境監測等方面[11-15]。目前對于煙草酯類香味成分的檢測主要基于非靶標GC/MS 內標相對定量方法,僅能測定可準確定性的化合物。受樣品基質干擾等問題制約,所檢測的指標一般低于10 種,且靈敏度、通量均不能滿足實際應用的需求。而GC-MS/MS 技術[16]具有多靶標、高通量、高靈敏度、日間平行性好、線性范圍寬等優點,目前已廣泛應用于農藥殘留、食品風味檢測及環境監測[17-20]等方面。
本研究中通過對國內外煙草添加劑名單、煙草成分分析文獻的整合、梳理,篩選出57 種對煙草感官品質有影響的關鍵酯類香味成分。通過對樣品前處理、色譜-質譜條件優化,建立了酸性條件泡發、乙腈提取、無水MgSO4除水、GC-MS/MS 同時測定煙草中57 種關鍵酯類香味成分的分析方法,并建立標準曲線對這些成分的標準樣品進行絕對定量,使用t 檢驗和偏最小二乘法判別分析(PLS-DA)篩選出差異成分并成功區分出不同風格的卷煙,旨在為煙草酯類香味成分分析提供方法參考。
1 材料與方法
1.1 材料、試劑和儀器
煙末(2016 年云南曲靖產C3F);15 種市售商品卷煙:1#~5#為清香風格卷煙,6#~10#為苔香風格卷煙,11#~15#為焦甜風格卷煙。
QuEChERS萃取試劑盒(4 g MgSO4+1 g NaCl)、分散固相萃取試劑盒(50 mg PSA(N-丙基乙二胺)+150 mg MgSO4)(美國Agilent 公司);57 種酯類標準品(>98%,分別購自美國Sigma-Aldrich 公司、日本TCI 公司、北京百靈威科技有限公司);苯乙酮-d8(98 atom% D,美國Sigma-Aldrich 公司);乙腈(色譜純,美國J T Baker 公司);二氯甲烷(色譜純,德國Chemicell 公司)。
Sin-QuEChERS Nano 簡單基質小柱(包括2 g Na2SO4、0.8 g MgSO4、90 mg PSA)、Sin-QuEChERS Nano 復雜基質小柱(包括2 g Na2SO4、0.6 g MgSO4、60 mg PSA、30 mg GCB(石墨化炭黑)和25 mg MWCNTs(多壁碳納米管))(北京綠綿科技有限公司);7890B-7000D 氣相色譜/三重四極桿質譜聯用儀(美國Agilent 公司);EOFO-945066 多管式旋渦混合器(美國Talboys 公司);3-30KS 高速冷凍離心機(德國Sigma公司);Milli-Q超純水儀(美國Millipore公司)。
1.2 樣品前處理
稱取2 g 煙末樣品于50 mL 具塞離心管中,加入10 mL 磷酸鹽緩沖液,調pH 為3,渦旋使樣品完全浸潤,靜置5 min;加入10 mL乙腈以及80 μL 30.0 mg/L苯乙酮-d8內標工作液,以2 500 r/min 渦旋10 min,而后放入-18 ℃的冰箱中冷凍10 min。取出后加入4 g 無水硫酸鎂、1 g 氯化鈉并迅速振搖,再以2 500 r/min 渦旋2 min,7 000 r/min 離心3 min;取1.5 mL 上清液,加入0.2 g 無水硫酸鎂,立即以2 500 r/min渦旋2 min,7 000 r/min 離心3 min;然后將上清液過0.22 μm 有機相濾膜后進行GC-MS/MS 檢測。檢測條件:
色譜柱:DB-5MS UI 彈性石英毛細管色譜柱(60 m×0.25 mm×0.25 μm),進樣口端串聯預柱(5 m×0.25 mm);進樣口溫度:280 ℃;程序升溫:75 ℃(5 min)進樣模式:不分流進樣,不分流時間1 min;載氣:氦氣(99.999%),恒流模式,流速1.5 mL/min;進樣量:0.8 μL。電離模式:電子轟擊(EI);電離能:70 eV;燈絲電流:35 μA;離子源溫度:230 ℃;四極桿溫度:150 ℃;傳輸線溫度:280 ℃;Q2碰撞氣:氮氣(99.999%),流量1.5 mL/min;淬滅氣:氦氣(99.999%),流量2.25 mL/min;掃描方式:多反應監測(dMRM)。MassHunter 工作站用于儀器控制和數據處理。
2 結果與討論
2.1 酯類化合物及內標化合物的篩選
通過對煙草添加劑以及煙草成分分析文獻的整合、梳理及篩選,選擇對感官質量影響較大的57 種酯類化合物,如表1 所示。
2.2 質譜條件的優化
需要優化的質譜參數主要有母離子、子離子和碰撞能量。首先將各化合物進行全掃描(Full scan)分析(m/z 20~330),確定保留時間及一級質譜圖,并篩選2~4 個質荷比(m/z)及豐度較大的離子作為備選母離子;再將上述各母離子在不同的碰撞能量(5、10、15、20、25、30、35、40 eV)下進行產物離子掃描(Product ion scan),每個化合物篩選出4~8 對離子對及最優碰撞能量;最后,用選擇多反應監測模式(dMRM)分析標準溶液、基質提取液、添加標準品的提取液,選擇抗干擾能力強、靈敏度高的兩對離子對分別作為定量及定性離子對。57 種酯類化合物及內標的保留時間、母離子、子離子及碰撞能量見表1,其中二氫茉莉酮酸甲酯具有順式、反式2 種構型,在結果中表現出2 個色譜峰、2 個保留時間。
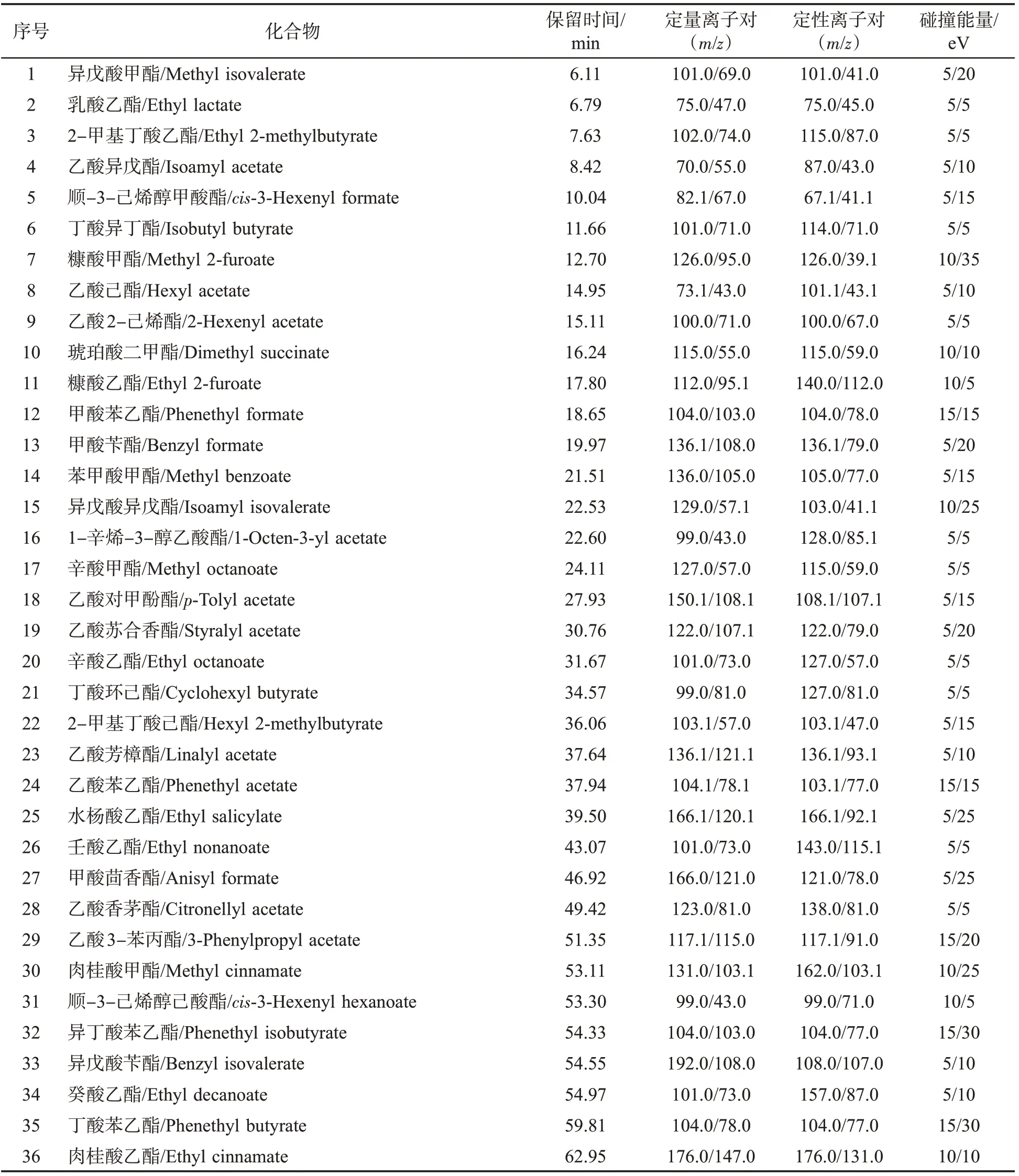
表1 57 種酯類化合物及其內標的MRM 參數Tab.1 MRM parameters of 57 esters and internal standard
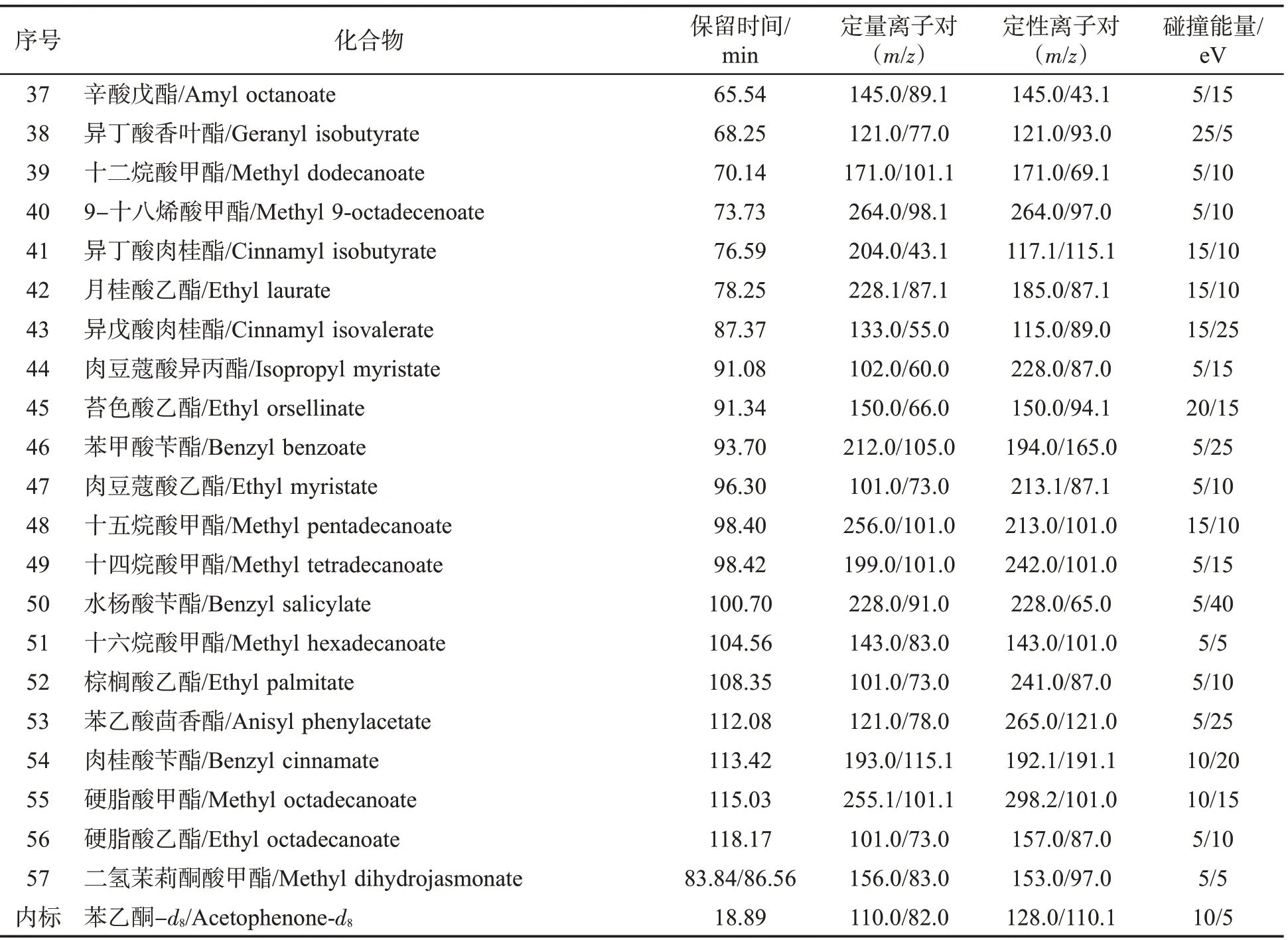
表1 (續)
2.3 提取方式的選擇
煙草香味成分的提取目前多采用同時蒸餾萃取(SDE)法,而QuEChERS 法具有簡便、高效等優點。以提取效率為標準,對比了同時蒸餾萃取法與QuEChERS法兩種前處理方式,并預先對同時蒸餾萃取法進行了優化。同時蒸餾萃取具體操作步驟如下:
稱取10 g 煙末,加入10 mL 泡發液(含10 mL H2O、0.55 g一水合檸檬酸以及3 g NaCl)泡發30 min,加入120 mL二氯甲烷(含3.045 μg/g 苯乙酮-d8內標)超聲0.5 h,過濾至1 000 mL 平底燒瓶中;將裝有提取液的1 000 mL燒瓶裝在同時蒸餾萃取裝置的一端,放入60 ℃水浴鍋中,另一端裝入100 mL 平底燒瓶,同時蒸餾萃取1 h。將100 mL平底燒瓶端放入60 ℃水浴鍋中;將裝有350 mL H2O 和90 g NaCl 的1 000 mL 平底燒瓶放入電熱套中,調節電熱套功率為0.3 kW,同時蒸餾萃取1.5 h,待冷卻后取下100 mL 平底燒瓶,加入無水Na2SO4干燥過夜。在常壓,水浴40 ℃的條件下旋轉蒸發濃縮至1~2 mL。過0.22 μm 有機相濾膜,進行GC-MS/MS 分析。
分別使用同時蒸餾萃取法和QuEChERS 法對樣品煙末進行提取,GC-MS/MS 法檢測,結果如圖1 所示。使用QuEChERS 法和同時蒸餾萃取法分別提取出14 和13 種化合物,差異化合物為丁酸苯乙酯。共同提取出的13 個化合物中,以可提取出化合物的絕對響應計,QuEChERS 法和同時蒸餾萃取法的提取結果絕對響應較高的分別有6 和7 種。其中異丁酸香葉酯、水楊酸芐酯、苯乙酸茴香酯、甲酸芐酯及乙酸苯乙酯5 種酯類QuEChERS 法的提取效率顯著高于同時蒸餾萃取法。同時蒸餾萃取法在提取時溫度較高,有三丁胺等副產物產生,并且操作繁瑣,耗時較長;而QuEChERS 提取法提取條件溫和,易于大批量制備,耗時短。因此,選擇QuEChERS 提取法作為前處理方法。
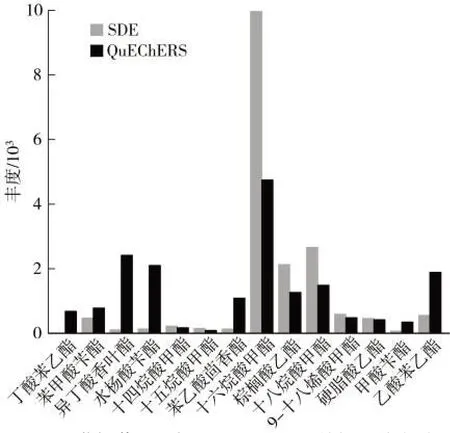
圖1 同時蒸餾萃取法與QuECHERS 法的提取效率對比Fig.1 Comparison of extraction efficiencies between SDE and QuECHERS methods
2.4 提取溶劑的選擇
文獻報道的煙草中香味成分檢測的提取溶劑多為二氯甲烷,乙腈在多組分分析中應用較為廣泛。因此本研究中以目標化合物在實際樣品中的提取效率為指標,考察了二氯甲烷和乙腈兩種溶劑。如圖2 所示,用乙腈作為提取溶劑時,共提取出14 種目標物;而二氯甲烷提取液中僅檢測出9 種酯類目標物,丁酸苯乙酯、苯甲酸芐酯、異丁酸香葉酯、水楊酸芐酯及甲酸芐酯均未檢出。除十八烷酸甲酯和硬脂酸乙酯外,同一目標物在乙腈提取物中的濃度明顯高于二氯甲烷提取物。結果表明,乙腈的提取效率顯著高于二氯甲烷。并且乙腈作為提取溶劑時,色譜峰型優于二氯甲烷,3 次平行實驗的相對標準偏差更小,實驗平行性更好。因此,選擇乙腈作為提取溶劑。
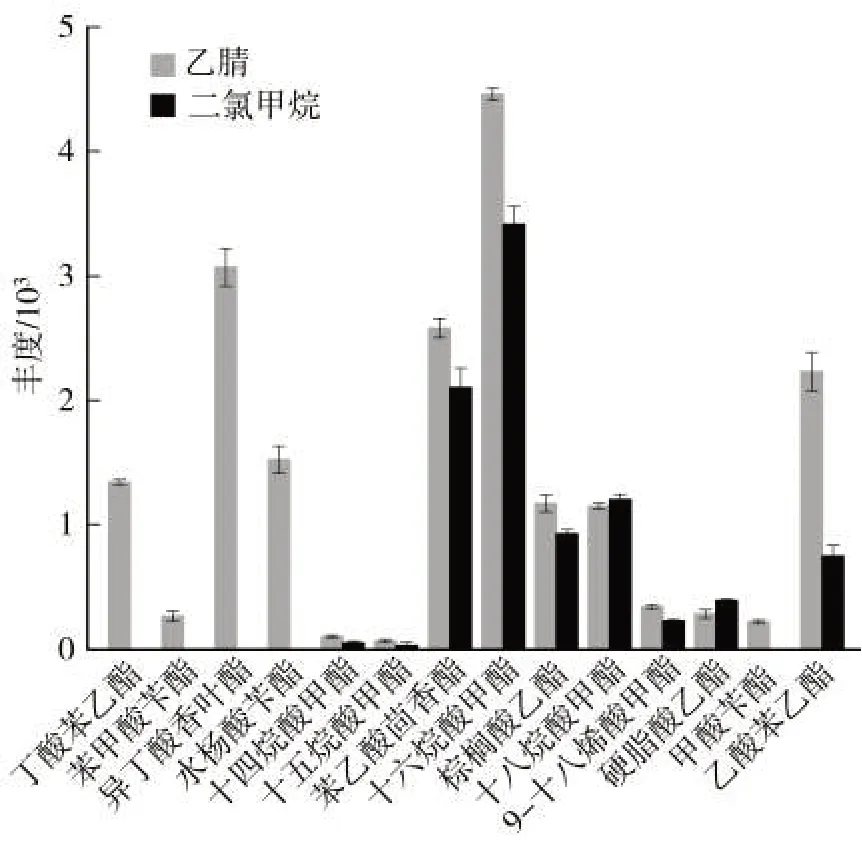
圖2 二氯甲烷與乙腈的提取效率對比(n=3)Fig.2 Comparison of extraction efficiencies between dichloromethane and acetonitrile(n=3)
2.5 泡發液pH 的優化
煙草中含有大量煙堿,提取液中煙堿濃度過高會降低色譜柱柱效,并導致煙堿附近出峰物質的保留時間漂移。因此,擬采用降低pH的方式降低提取液中煙堿濃度以消除其對目標物檢測的影響。樣品煙末經超純水泡發后pH 約為5.6。使用磷酸及磷酸二氫鈉鹽緩沖液將樣品煙末泡發后pH 分別調節至3.0 和4.0。考察了不同pH條件下各目標物的提取效果以及提取液中煙堿濃度的差異。由于煙堿濃度過大,圖3 中的響應值為其實際響應的十萬分之一。pH為4.0 及5.6 時,煙堿濃度極高,pH為3.0 時其濃度顯著降低,僅是pH 為5.6 時的3%左右。煙葉樣品中可檢測到的14 種酯類化合物中,丁酸苯乙酯、異丁酸香葉酯、水楊酸芐酯和乙酸苯乙酯在pH 為3.0 時的提取效率明顯優于pH 為4.0 及5.6 時,苯乙酸茴香酯和十六烷酸甲酯在pH為3.0時的提取效率不及pH為4.0 和5.6 時,但其響應較高,pH 為3.0 時仍能滿足檢測需要,其余化合物在不同pH時的提取效率無顯著性差異。因此,選擇在pH 為3.0 時進行泡發。
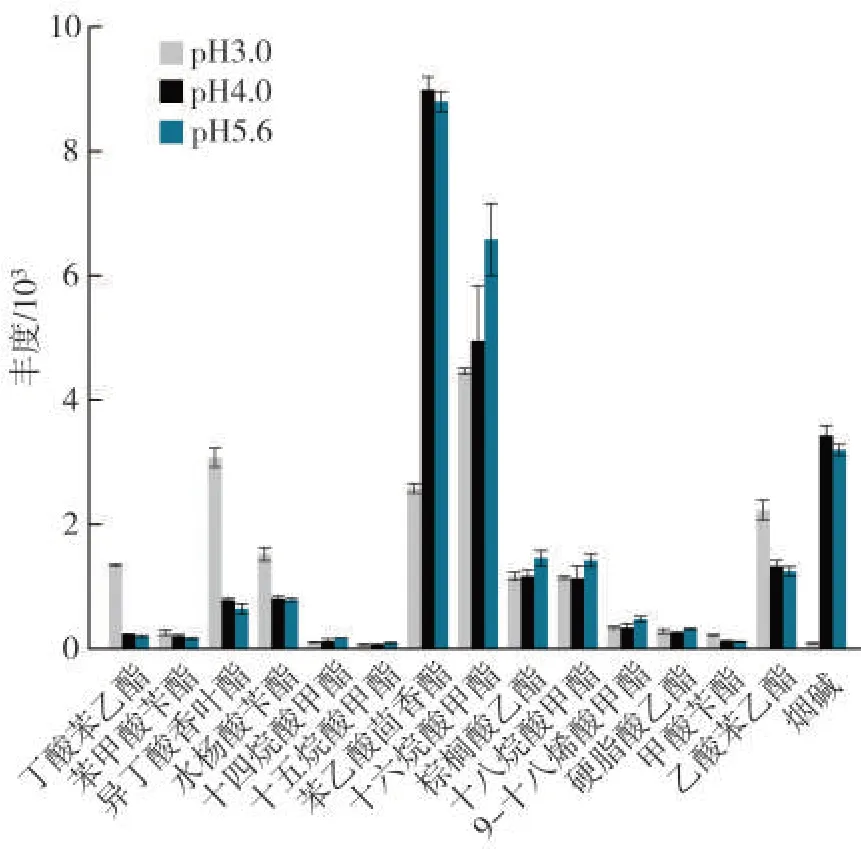
圖3 不同泡發pH 對提取效率的影響(n=3)Fig.3 Effects of soaking pH value on extraction efficiency(n=3)
2.6 泡發時間的選擇
在pH 為3.0 條件下考察了不同泡發時間對實際樣品中目標物提取效率的影響,時間分別為0(即不泡發)、5、10、20、30 min,其對各目標物最終的提取效率如圖4 所示。結果表明,泡發時間對提取效率沒有明顯影響。然而不泡發時,提取液中含有較多煙堿,不利于后續檢測。因此,選擇泡發提取效率相對較高、用時相對較短的泡發時間,即泡發5 min。

圖4 不同泡發時間對提取效率的影響(n=3)Fig.4 Effects of soaking time on extraction efficiency(n=3)
2.7 渦旋時間的選擇
考察了渦旋時間分別為2、5、10、20、30 min 時對實際樣品的提取效率,結果如圖5 所示。苯乙酸茴香酯、十六烷酸甲酯、棕櫚酸乙酯、十八烷酸甲酯、9-十八烯酸甲酯隨渦旋時間延長,提取效率有增加的趨勢;丁酸苯乙酯、異丁酸香葉酯、水楊酸芐酯隨提取時間延長,提取效率降低;其余目標物的提取效率隨提取時間延長變化趨勢不明顯。綜合以上結果,渦旋時間選擇為10 min。
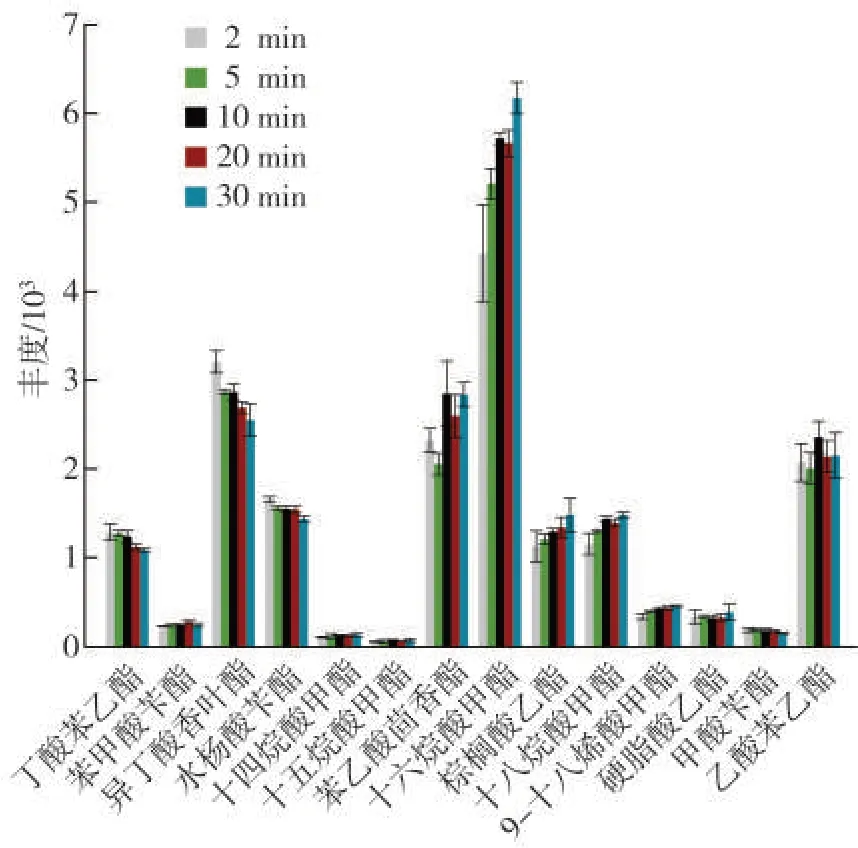
圖5 不同渦旋時間對提取效率的影響(n=3)Fig.5 Effects of vortexing time on extraction efficiency(n=3)
2.8 凈化劑的選擇
考察了4 種不同凈化方式對煙草樣品提取液的凈化效果及對目標物的吸附效應:取1.5 mL 上清液于離心管中,加入200 mg 無水硫酸鎂或50 mg PSA和150 mg 無水硫酸鎂;分別取Sin-QuEChERS Nano簡單基質小柱、Sin-QuEChERS Nano 復雜基質小柱,置于有提取液的50 mL 離心管中,緩慢下壓至刻度處,吸取上清液,過0.22 μm有機相濾膜。結果表明,若提取液不凈化,含水率約為10%;加入150 mg 無水硫酸鎂后,含水率降為3%,對儀器更加友好。圖6 為各方式凈化后目標化合物的響應值。可以看出,用PSA凈化時,對部分目標物吸附嚴重,丁酸苯乙酯、異丁酸香葉酯均無法檢出,除苯乙酸茴香酯外,其他目標物均有不同程度吸附;用Sin-QuEChERS Nano簡單基質小柱凈化時,苯甲酸芐酯、異丁酸香葉酯、十五烷酸甲酯吸附嚴重,無法檢出,除苯乙酸茴香酯外,其他目標物均有不同程度吸附;而使用Sin-QuEChERS Nano 復雜基質小柱凈化時,苯甲酸芐酯、異丁酸香葉酯、水楊酸芐酯、十四烷酸甲酯、十五烷酸甲酯、9-十八烯酸甲酯均無法檢出,其余均有不同程度吸附。因此,最終使用無水MgSO4除水。
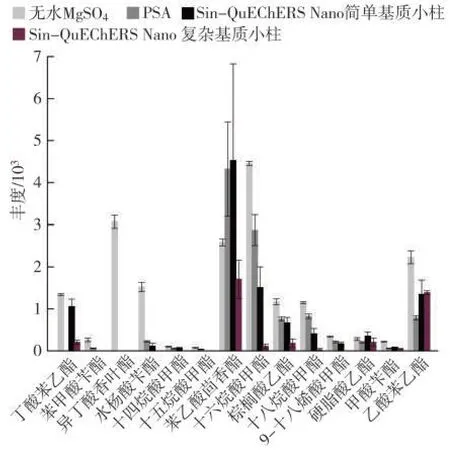
圖6 不同凈化方式對提取效率的影響(n=3)Fig.6 Effects of purification methods on extraction efficiency(n=3)
2.9 基質效應
本研究中通過公式(1)考察了57 種酯類香味成分的基質效應:

式中:A 為溶劑標準工作溶液標準曲線的斜率;B 為基質匹配標準工作溶液標準曲線的斜率。
基質效應越接近1,說明基質效應越不明顯。結果表明,11種目標物的基質效應均不明顯(0.9 <ME <1.1);有24 種目標物存在基質減弱效應(ME <1.0),33 種目標物存在基質增強效應(ME >1.0),其中苯甲酸芐酯、肉桂酸芐酯、苯乙酸茴香酯、9-十八烯酸甲酯、苔色酸乙酯5 種酯類化合物存在明顯的基質效應,使用純溶劑標準工作溶液繪制標準曲線時,除高濃度點外,較低濃度點無信號響應,但其基質匹配標準工作溶液線性關系良好(表2)。因二氫茉莉酮酸甲酯有兩個峰,故總共有58 個色譜峰。因此,采用基質匹配標準工作液以校正基質效應引入的定量誤差。
對比河南、廣西、黑龍江及云南4 個產地的煙葉表明,云南煙葉(C3F,2016年)含有較少的目標成分,因此選擇云南煙葉提取液作為基質空白提取液。基質匹配標準曲線配制方法:預先使用乙腈配制混合母液(100 mg/mL),而后以基質空白提取液作為稀釋劑配制目標濃度的工作液。
2.10 方法評價
為了減少基質效應的影響,采用基質匹配標準工作溶液繪制標準曲線,標準工作溶液濃度為10、20、50、100、200、500、1 000 ng/mL。其中有6 種化合物由于基質中質量分數較高等原因,標準曲線繪制范圍不同。十八烷酸甲酯、9-十八烯酸甲酯、乙酸己酯和丁酸環己酯4 種化合物,無10 ng/mL 點;甲酸苯乙酯無10、20 ng/mL點;十六烷酸甲酯標準工作溶液的濃度為100、200、500、1 000、2 000、5 000、10 000、20 000 ng/mL。考察0.05、0.50、5.00 mg/kg 3 個水平的加標回收率,以10 倍信噪比作為方法定量限。如表3 所示,各化合物均有良好的線性關系(r2>0.99),平均回收率范圍為62.5%~128.7%,RSD 在0.5~25.4%之間。以平均回收率為60%~120%且RSD<20%為標準,在低添加水平下,47 個化合物達到標準;在中、高添加水平下,57個化合物全部達到標準。定量限為3~74 μg/kg,其中有50個化合物的定量限≤50 μg/kg。另由于二氫茉莉酮酸甲酯具有2 個異構體,其方法學性質不同,分別列出。該方法具有良好的精密度、靈敏度,可滿足分析檢測需要。

表2 57 種酯類化合物在煙草提取液中的基質效應Tab.2 Matrix effects of 57 esters in tobacco extract
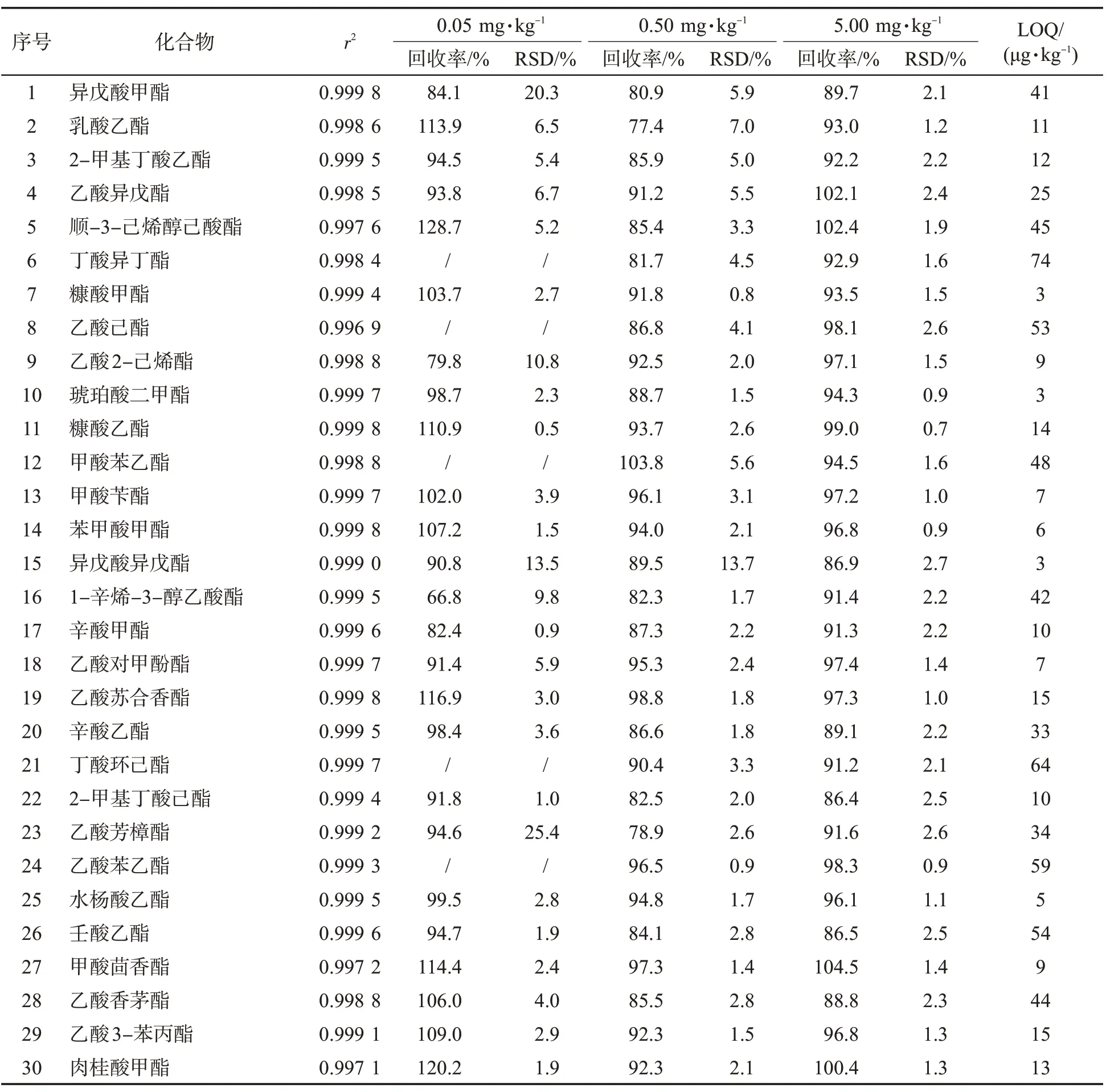
表3 57 種酯類化合物的相關系數、回收率(n=5)、相對標準偏差和定量限Tab.3 Correlation coefficients, recoveries (n=5), relative standard deviations (RSDs)and limits of quantification (LOQs) of 57 esters
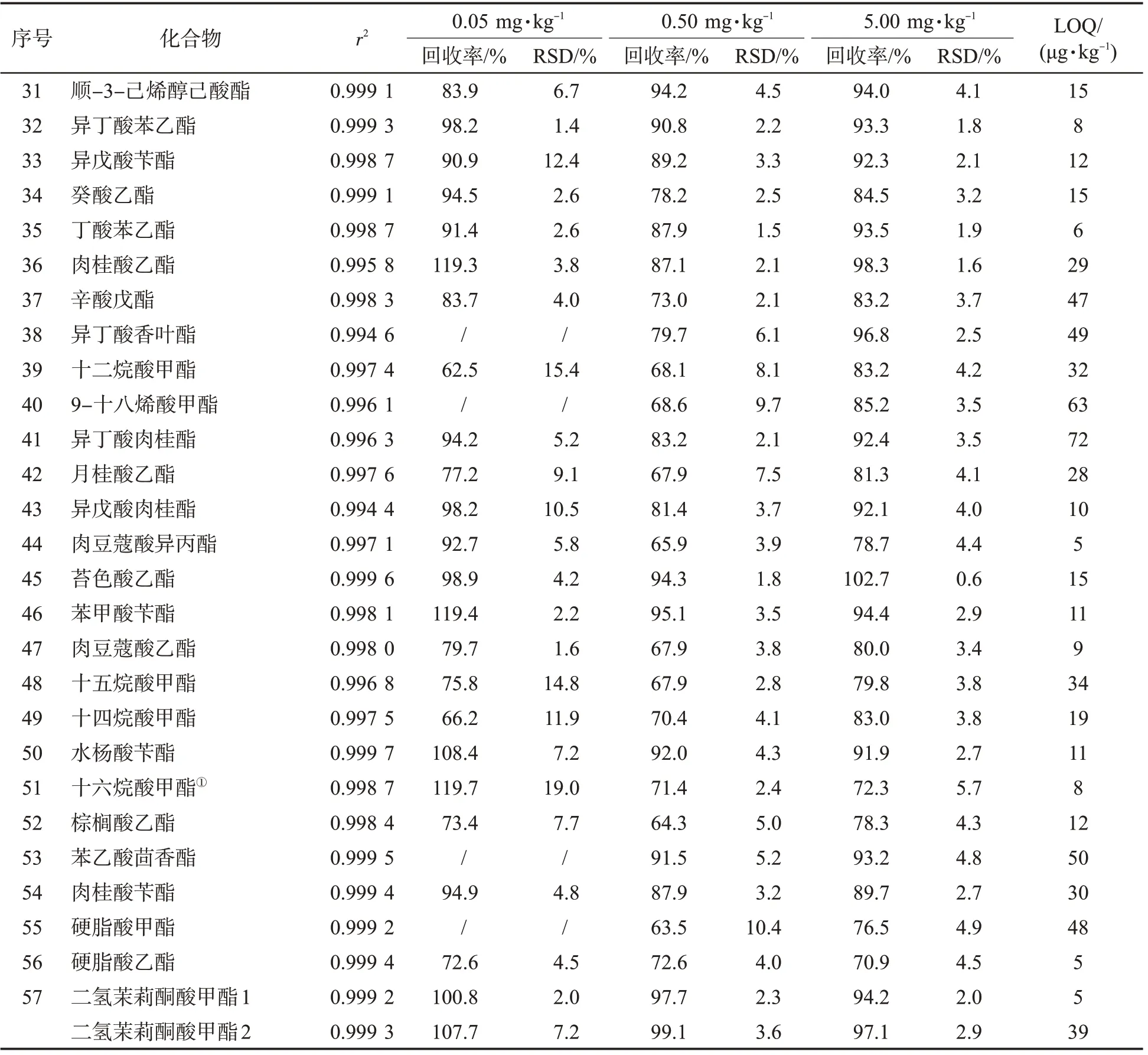
表3 (續)
2.11 實際樣品檢測結果分析
用該方法檢測了15 個不同風格的卷煙樣品,共檢出了31 種酯類化合物,其中十六烷酸甲酯的質量分數最高,其次是十八烷酸甲酯、棕櫚酸乙酯和9-十八烯酸甲酯,見表4。在可檢出的化合物中,以P值小于0.05 為標準,使用t 檢驗篩選出8 種具有顯著性差異的成分,如表5 所示。使用最小二乘法判別分析該8 種酯類,結果見圖7。可知,不同風格的卷煙呈顯著性差異,其中苔香風格與焦甜風格組內差異較小,清香風格組內風格差異較大。

表4 15 種市售卷煙的檢測結果Tab.4 Detection results of 15 commercial cigarette samples
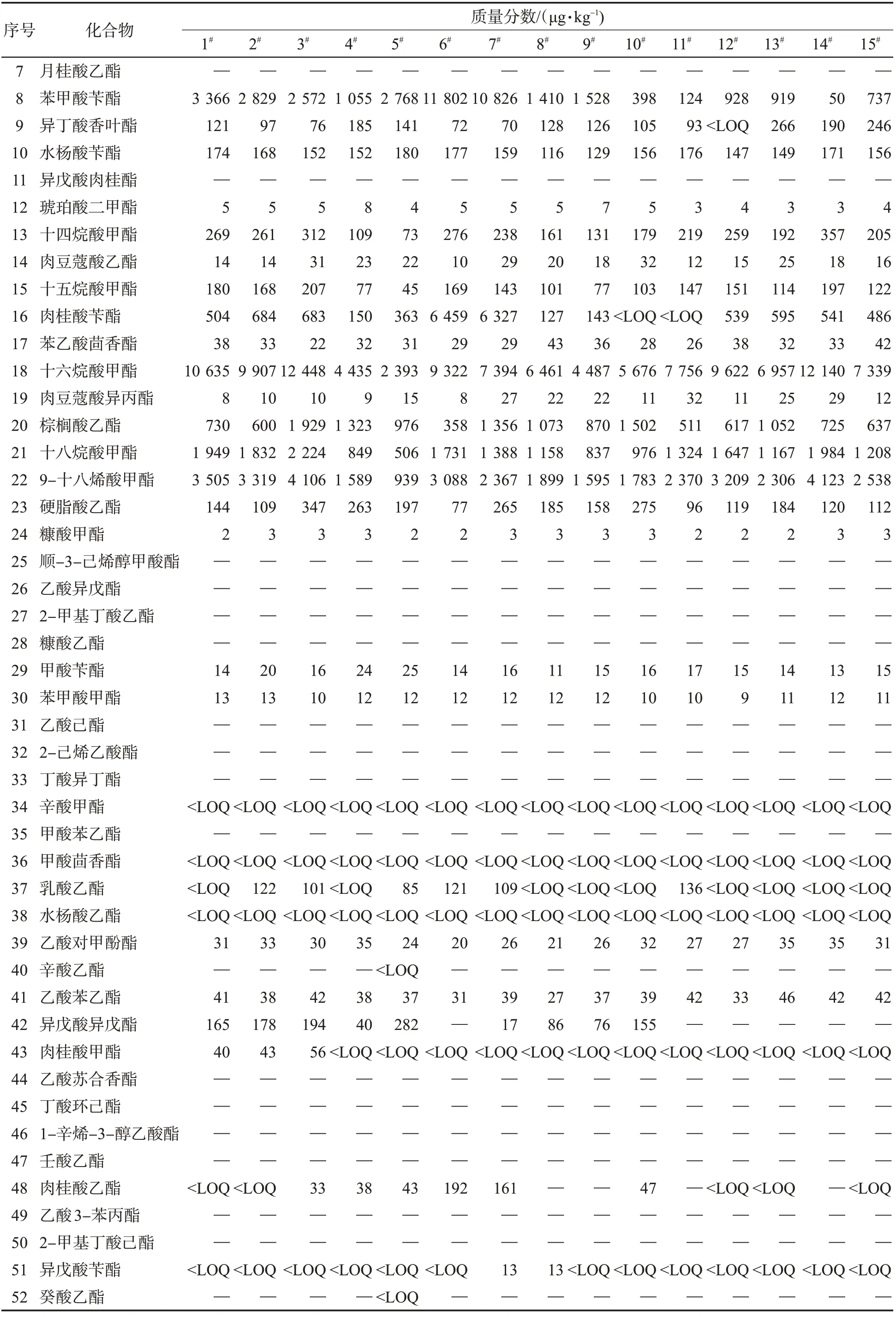
表4 (續)
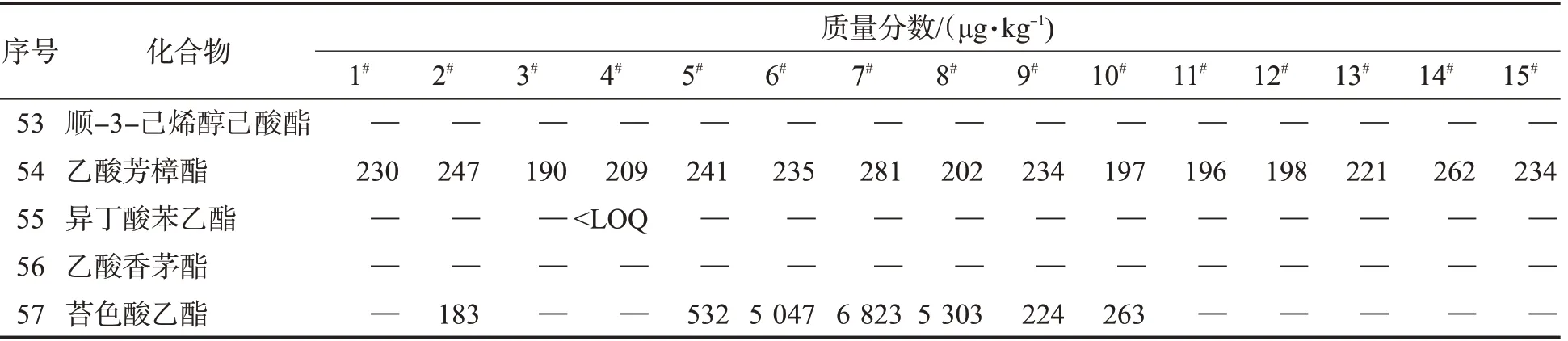
表4 (續)
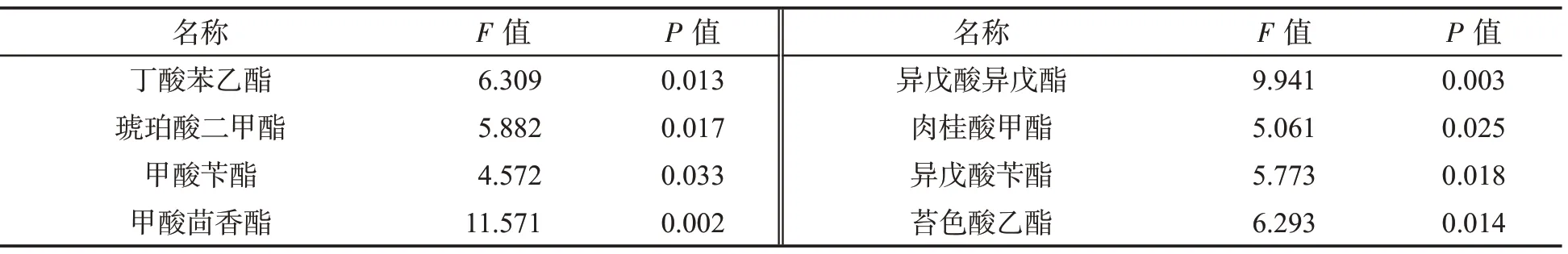
表5 8 種具有顯著性差異的酯類香味成分的P 值及F 值Tab.5 P and F values of 8 esters with significant differences

圖7 8 種具有顯著性差異的酯類香味成分的PLS-DA 分析結果Fig.7 Results of PLS-DA analysis of 8 esters with significant differences
3 結論
①通過優化的QuEChERS 前處理方法結合GC-MS/MS 技術建立了檢測煙草中重要酯類香味成分的方法,能滿足煙草樣品中57 種對感官品質有影響的酯類香味成分檢測需求。②對于實際樣品檢測,可以較好地區分不同風格的卷煙樣品,證明所建立的方法具有良好的實用性。③與傳統方法相比,本方法在靶標物數量、靈敏度和精密度方面顯著提高,具有簡便、高效、高通量等優點。
猜你喜歡
奧秘(創新大賽)(2023年3期)2023-05-06 01:48:20
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
甘肅教育(2020年14期)2020-09-11 07:57:42
浙江中西醫結合雜志(2017年2期)2017-01-12 18:23:59
海峽科技與產業(2016年3期)2016-05-17 04:32:12
當代化工研究(2016年9期)2016-03-20 16:22:08
時代英語·高二(2015年1期)2015-03-16 00:08:11
中國衛生(2014年11期)2014-11-12 13:11:32
