茶皂素的分離純化方法的初步研究
2019-12-10 09:02:16湛詠詩廖華衛(wèi)周雄堅
科技資訊 2019年28期
湛詠詩 廖華衛(wèi) 周雄堅
摘? 要:目的 研究油茶餅粕中茶皂素的分離純化方法。方法 采用紫外分光光度法對萃取法、薄層色譜分離法及柱層析法進(jìn)行跟蹤檢測。結(jié)果 萃取后樣品純度35%,薄層板制備后樣品純度60%,柱層析后樣品純度86%。結(jié)論 茶皂素的初步研究效果較佳。
關(guān)鍵詞:茶皂素? 分離純化? 薄層色譜? 柱層析
中圖分類號:Q946 ? ?文獻(xiàn)標(biāo)識碼:A 文章編號:1672-3791(2019)10(a)-0032-02
Abstract: Objective: To study the separation and purification of tea saponin from Camellia oleifera. Methods:Ultraviolet spectrophotometry was used to detect the extraction method, thin layer chromatography and column chromatography. Results:The purity of the sample after extraction was 35%. The purity of the sample after preparation of the thin layer plate was 60%, and the purity of the sample after column chromatography was 86%. Conclusion: The preliminary study of tea saponin has better effect.
Key Words: Tea saponin; Separation and purification; Thin layer chromatography; Column chromatography
油茶籽榨取茶油后剩下的渣餅即為油茶餅粕,是茶油生產(chǎn)的下腳料茶皂素又名茶皂甙,是一種優(yōu)良的非離子型天然表面活性劑,具有溶血、抗菌、抗病毒、抗應(yīng)激、降血脂等多種生理活性,可廣泛應(yīng)用于日化、醫(yī)藥、食品和農(nóng)藥等行業(yè)。茶皂素是一類結(jié)構(gòu)相似的齊墩果烷型三萜類皂苷混合物,其單體結(jié)構(gòu)稱為茶皂苷,由皂苷元、糖體、有機(jī)酸(或糖醛酸)3個部分組成。茶皂素為乳白色或淡黃色無定形粉末,平均分子式為 C57H90O26,相對分子量為1200~2800左右,熔點(diǎn)223℃~224℃,是一種非離子型極性物質(zhì)[1],該實(shí)驗(yàn)采取萃取、TLC色譜法、柱層析法等分離純化方法,為建立茶皂素的分離純化方法提供參考依據(jù)。
1? 儀器與試劑
儀器:紫外可見分光光度計UV-6100S。
試劑:硫酸、香草醛、無水乙醇、乙酸乙酯、正丁醇等均為分析純茶皂素對照品(B20145,上海源葉生物科技有限公司)
2? 方法與結(jié)果
2.1 油茶枯預(yù)處理及茶皂素的提取
2.1.1 油茶枯預(yù)處理
取油茶枯1kg,粉碎,于60℃條件下干燥4h,控制水分<5%,粉碎過60目篩,然后加一定量的石油醚(30~60),經(jīng)索氏抽提至提取液無色,除去油脂。將除去油脂后的茶籽粕置于通風(fēng)良好處使其自然風(fēng)干,然后放入60℃烘干,備用。
2.1.2 茶皂素的提取
取2.1.1中油茶枯預(yù)處理好的樣品100g置于2000mL的三角瓶中,按1∶10的比例加入50%乙醇,超聲波提取,每次2h,提取3次。抽濾,濾液合并,回收乙醇,濾液水浴濃縮至200g即可(備用)。
2.2 茶皂素含量測定方法
2.2.1 標(biāo)準(zhǔn)曲線
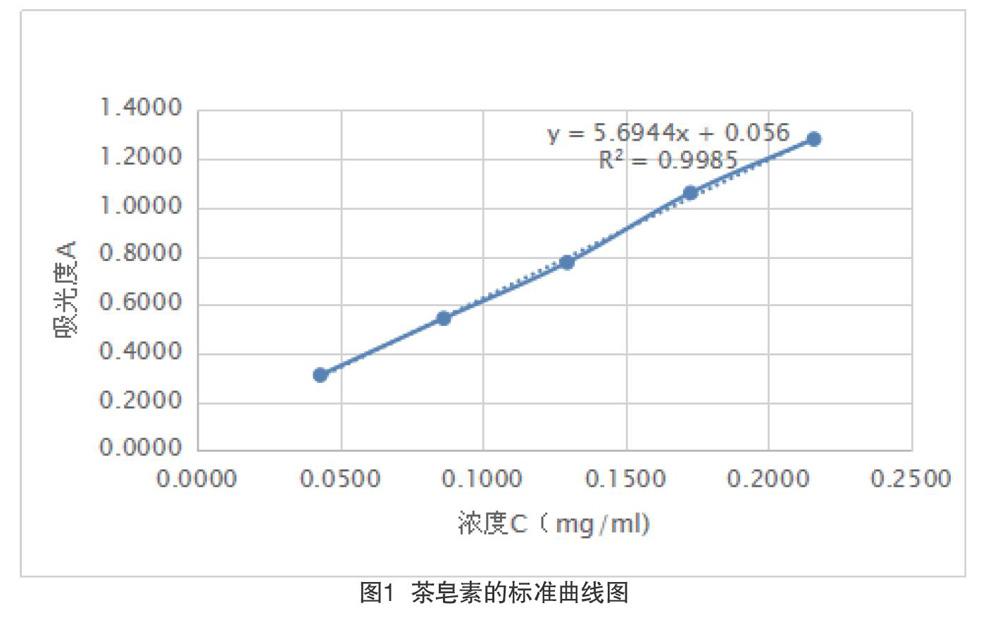
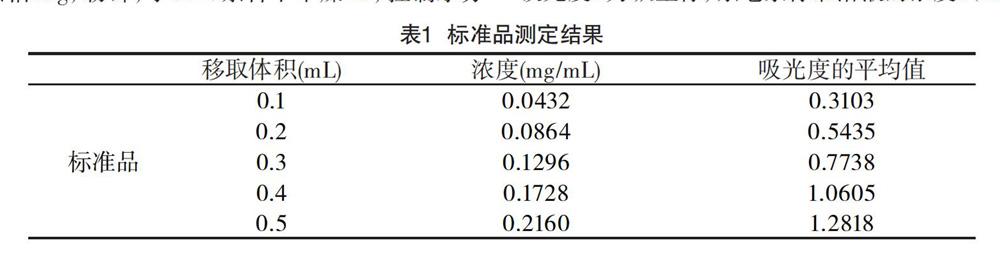
取一個20mL容量瓶準(zhǔn)確配制濃度2mg/mt的茶皂素標(biāo)準(zhǔn)溶液備用,用移液槍精確移取茶皂素標(biāo)準(zhǔn)溶液0.1、0.2、0.3、0.4、0.5mL,分別放置于5個比色管中,并加水至溶液體積均為0.5mL。精密加入8%香草醛95%乙醇溶液0.5mL,將比色管置于冰水混合物中,移取77%的濃硫酸4mL加入比色管中,振蕩后于60℃的水浴鍋中水浴15min,然后將比色管放置于冰水混合物中反應(yīng)10min,取出放置至室溫,以試劑空白為對照,在其最大吸收波長550nm處測定其吸光度,以吸光度A為縱坐標(biāo),茶皂素標(biāo)準(zhǔn)溶液的濃度c(mg/mL)為橫坐標(biāo),得到線性標(biāo)準(zhǔn)方程。
2.2.2 含量測定
取提取液0.5mL,置于比色管中,精密加入8%香草醛乙醇溶液0.5mL,將比色管置于冰水混合物中,移取77%的濃硫酸4mL加入比色管中,振蕩后于60℃的水浴鍋中水浴15min,然后將比色管放置于冰水混合物中反應(yīng)10min,取出使其恢復(fù)室溫,以試劑空白為對照,在其最大吸收波長550nm處測定其吸光度[2],依據(jù)2.2.1標(biāo)準(zhǔn)曲線計算含量。
2.3 樣品液的萃取
將上述溶液置于1000mL的分液漏斗中,先用石油醚(60~90)萃取,每次200mL,共3次,合并萃取液,濃縮(備用)。再用乙酸乙酯萃取,每次200mL,共3次,合并萃取液,濃縮(備用)。最后用正丁醇萃取,每次200mL,共3次,合并萃取液,濃縮至干(備用)。
2.4 TLC色譜法
將上述正丁醇萃取部位的樣品用熱乙醇溶解,每次30mL,共3次,合并溶液。濃縮轉(zhuǎn)移至10mL容量瓶中作為樣品溶液(備用)。將樣品溶液點(diǎn)于已活化好的10cmx10cm的薄層板上,在1.5cm處帶狀點(diǎn)樣,在相應(yīng)的位置點(diǎn)茶皂素對照品溶液,以乙酸乙酯-乙醇(1∶4)為展開劑展開,展開至約8.5cm處即可,取出,晾干,碘熏顯色,用鉛筆做好記號,取出,記號范圍適當(dāng)擴(kuò)大,將吸附有樣品的吸附劑按色帶的位置刮下,收集好,置于層析柱上,用熱乙醇洗脫,收集洗脫液,濃縮至10mL(備用)。
2.5 柱層析分離純化
將10g柱層析硅膠(100~200目)加入到上述溶液中,置于蒸發(fā)皿中,攪拌均勻,水浴蒸干,80℃烘干,備用。稱取300g已經(jīng)活化好的柱層析硅膠(100~200目),以乙酸乙酯-乙醇(4∶1)裝柱,裝好柱后用洗脫劑洗脫一天,干法上樣,上好樣品后在硅膠上加一團(tuán)脫脂棉,打開活塞,進(jìn)行洗脫,收集流份,每瓶約20mL,薄層色譜檢測,合并茶皂素流份,回收洗脫劑,水浴蒸干,熱乙醇適量溶解,濾過,放冷析晶。濾過,收集結(jié)晶,烘干,依樣品測定方法測定含量。濾液也依樣品測定方法測定含量。觀察各自的含量情況,從而為摸索一個更好地分離純化方法提供參考。
3? 討論
此次實(shí)驗(yàn)摸索了茶皂素的分離純化方法,每一步都進(jìn)行了跟蹤檢測,總體了解了茶皂素的占比情況,為以后進(jìn)一步的研究提供技術(shù)支撐。此次實(shí)驗(yàn)可操作性強(qiáng),容易學(xué)習(xí)領(lǐng)會,通俗易懂,萃取過程中,通過收集3種萃取溶液,分別跟蹤檢測,得到乙酸乙酯部位茶皂素占比約10%,正丁醇部位茶皂素占比約71%,剩余母液茶皂素占比約6%;薄層色譜制備中,展開系統(tǒng)選擇很重要,層析板鋪的好壞也對分離效果影響較大,跟茶皂素相應(yīng)部位的斑點(diǎn)處刮下后處理得到的茶皂素占比約75%,為了更深入地了解茶皂素的占比情況,斑點(diǎn)之上之下分別檢測,之上斑點(diǎn)處刮下的硅膠粉經(jīng)處理后檢測,經(jīng)檢測茶皂素占比約11%,之下斑點(diǎn)處刮下的硅膠粉經(jīng)處理后檢測,經(jīng)檢測茶皂素占比約4%;另外柱層析中,裝柱的好壞也是分離效果好壞的較為重要的因素。但回收率偏低,下一步努力的方向。設(shè)想如果每一步重復(fù)做多一次,估計會提高樣品的得率及純度。
參考文獻(xiàn)
[1] 鄧桂蘭.茶皂素的提取及純化研究[J].食品研究與開發(fā),2016,37(8):197-204.
[2] 傅春玲,洪奇華,阮輝,等.茶皂素定量測定方法的研究[J].杭州大學(xué)學(xué)報:自然科學(xué)版,1997,24(3):240-242.
