殼聚糖/鹽酸小檗堿緩釋微球的制備
2019-11-21 06:03:44林欣雨李瑤琦
中成藥 2019年11期
關鍵詞:殼聚糖
林欣雨,李瑤琦,周 瑾,羅 超
(紹興文理學院元培學院,浙江 紹興312000)
微球是指藥物分散或被吸附在高分子、聚合物基質中而形成的微粒分散體系,可提高穩定性,降低刺激性,控制釋放,并具有被動靶向性,是現代藥物傳遞系統的主要載體形式之一[1]。目前,制備微球的方法主要有相轉變法[2]、乳化交聯法[3]、噴霧干燥法[4]、膜乳化法等,其中膜乳化法以無機微孔膜(SPG)為介質,利用膜微孔分離和毛細管作用原理,通過在膜一側施加壓力使分散溶液透過膜孔,以微小液滴的形式分散在與其不相溶的溶液中而形成乳液[5],從而交聯固化形成微球,該方法反應條件溫和,操作簡便,易于規模化生產,重復性好。
殼聚糖是自然界存儲量僅次于纖維素的天然高分子多糖,由蝦、蟹等甲殼動物外殼的甲殼素脫乙酰化而得[6],具有良好的生物相容性、生物可降解性、生物粘附性,是組織工程材料和藥物緩控釋載體材料的主要基質成分[7-8],可制成微球[3]、納米粒[9]、水凝膠[10]、微凝膠[11]等多種不同特性的材料。殼聚糖微球可通過包覆[4]、吸 附[3]、共聚[12]等多種手段負載藥物,并具有理想的緩控釋性能、黏膜粘附性,可提高藥物在體液環境中的穩定性和生物利用度。本實驗采用SPG 膜乳化法制備殼聚糖/鹽酸小檗堿緩釋微球,并優化制備工藝,表征理化特性,研究緩釋性能,以期為相關研究提供依據。
1 材料
殼聚糖(脫乙酰度95%,分子量1.2×105,浙江澳興生物技術有限公司);鹽酸小檗堿原料藥(含有量>98%,鄭州豐冠化工產品有限公司);鹽酸小檗堿對照品(江蘇永健醫藥科技有限公司,批號200351-161107);司盤-80(上海薩恩化學技術有限公司)。戊二醛(東京仁成工業株式會社)。乙腈為色譜純;其余試劑均為分析純。
Waters e2695-2489型高效液相色譜儀(美國Waters 公司);Nicolet 740型紅外光譜儀(美國Thermo 公司);X’Pert PRO 型X 射線衍射儀(荷蘭PNAlytical 公司);JSM-6360LV 型掃描電子顯微鏡(日本電子株式會社);FD-1 A-50型冷凍干燥機(北京博醫康實驗儀器有限公司);高效手動膜乳化器(日本SPG Technology 公司)。
2 方法與結果
2.1 微球制備 將1 g 殼聚糖溶于50 mL 2%醋酸中,配制成2%溶液,在一定溫度下將200 mg 鹽酸小檗堿溶于其中,持續攪拌一定時間,作為水相,并以10倍體積液體石蠟-環己烷(7 ∶5)混合液為油相,2%司盤-80為乳化劑。在相同溫度下,通過50 μm SPG 膜乳化器將水相壓入到油相中,形成W/O 型乳液,再加入一定量戊二醛交聯1 h,離心收集微球,分別用乙醇、純水洗滌,真空冷凍干燥,即得。
2.2 含有量測定
2.2.1 色譜條件 SunFire C18色譜柱(4.6 mm×250 mm,5 μm);流動相乙腈-0.5% 磷酸(30 ∶70,三乙胺調pH 至3);體積流量1.0 mL/min;柱溫30 ℃;檢測波長345 nm;進樣量20 μL。
2.2.2 線性關系考察 精密稱取鹽酸小檗堿對照品10 mg,蒸餾水溶解并定容于100 mL 棕色量瓶中,配制成0.1 g/L,蒸餾水進一步稀釋至1.0、2.0、5.0、10.0、15.0、20.0 mg/L,在“2.1.1”項色譜條件下進樣測定。以峰面積積分值(Y)對溶液質量濃度(X)進行回歸,得方程為Y=6 139.2X-3 897.2(R2=0.999 9),在1~20 mg/L范圍內線性關系良好。
2.2.3 精密度試驗 1.0、10.0、20.0 mg/L 鹽酸小檗堿對照品溶液在“2.2.1”項色譜條件下進樣測定,在同1 d 內重復6次,測得鹽酸小檗堿峰面積RSD 分別為1.1%、2.0%、1.2%,表明儀器精密度良好。
2.2.4 穩定性試驗 10.0 mg/L 鹽酸小檗堿對照品溶液于0、2、4、6、8、12、24 h 在“2.2.1”項色譜條件下進樣測定,測得鹽酸小檗堿峰面積RSD 為1.1%,表明溶液在24 h 內穩定性良好。
2.2.5 加樣回收率試驗 配制含鹽酸小檗堿9.6、9.3、9.7、10.5、10.6、10.2 mg/L 的載藥微球溶出樣品溶液10 mL,加入對照品溶液(含10.0 mg/L鹽酸小檗堿)10 mL,在“2.2.1”項色譜條件下進樣測定,測得平均加樣回收率為99.12%,RSD為1.53%。
2.2.6 專屬性考察 制備鹽酸小檗堿對照品溶液、鹽酸小檗堿原料藥溶液、空白殼聚糖微球浸出液、載藥殼聚糖微球浸出液(釋放度檢測樣品),在“2.2.1”項色譜條件下進樣測定,色譜圖見圖1。由圖可知,載體材料對鹽酸小檗堿檢測沒有影響,該方法專屬性良好。

圖1 鹽酸小檗堿HPLC 色譜圖Fig.1 HPLC chromatograms of berberberine hydrochloride
2.3 包封率、載藥量測定 取適量載藥微球加到甲醇中,密封超聲1 h,10 000 r/min 離心30 min以使鹽酸小檗堿全部溶出,上清液加甲醇定容到100 mL,在“2.2.1”項色譜條件下進樣測定,按下式計算載藥量、包封率。

2.4 制備工藝優化[13]根據預實驗(單因素試驗)結果,選擇對載藥微球制備影響較大的交聯劑用量(X1)、混合攪拌時間(X2)、體系溫度(X3)作為影響因素,包封率(Y)作為評價指標,星點設計-效應面法進行優化,結果見表1。再通過Design-Expert 8.0.6.1軟件進行擬合,得到回歸方程為方差分析見表2。

表1 試驗設計與結果Tab.1 Design and results of tests
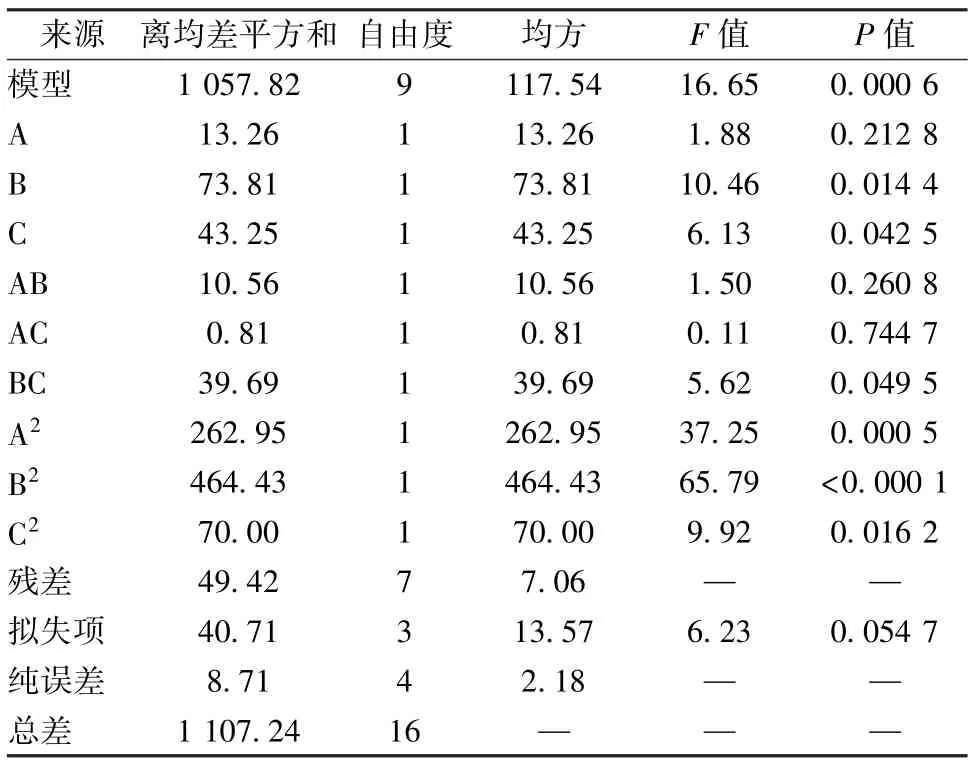
表2 方差分析Tab.2 Analysis of variance
由表2可知,模型P=0.000 6<0.05,表明其穩定可靠;復相關系數R2=0.955 4,表明模型擬合度良好,能準確預測實際情況;校正決定系數=0.898 0,表明模型能解釋89.80% 響應值的變化;各因素失擬項P>0.05,表明擬合方程準確性良好;各因素影響程度依次為X2>X3>X1,X2、X3、X2X3、有顯著影響(P<0.05)。
響應面分析見圖2。通過Design-Expert 8.0.6.1軟件,得到最優工藝為交聯劑用量8.23 mL,混合攪拌時間111.63 min,體系溫度43.72 ℃,包封率82.00%,考慮到實際可操作性,將其修正為交聯劑用量8 mL,混合攪拌時間112 min,體系溫度44 ℃。
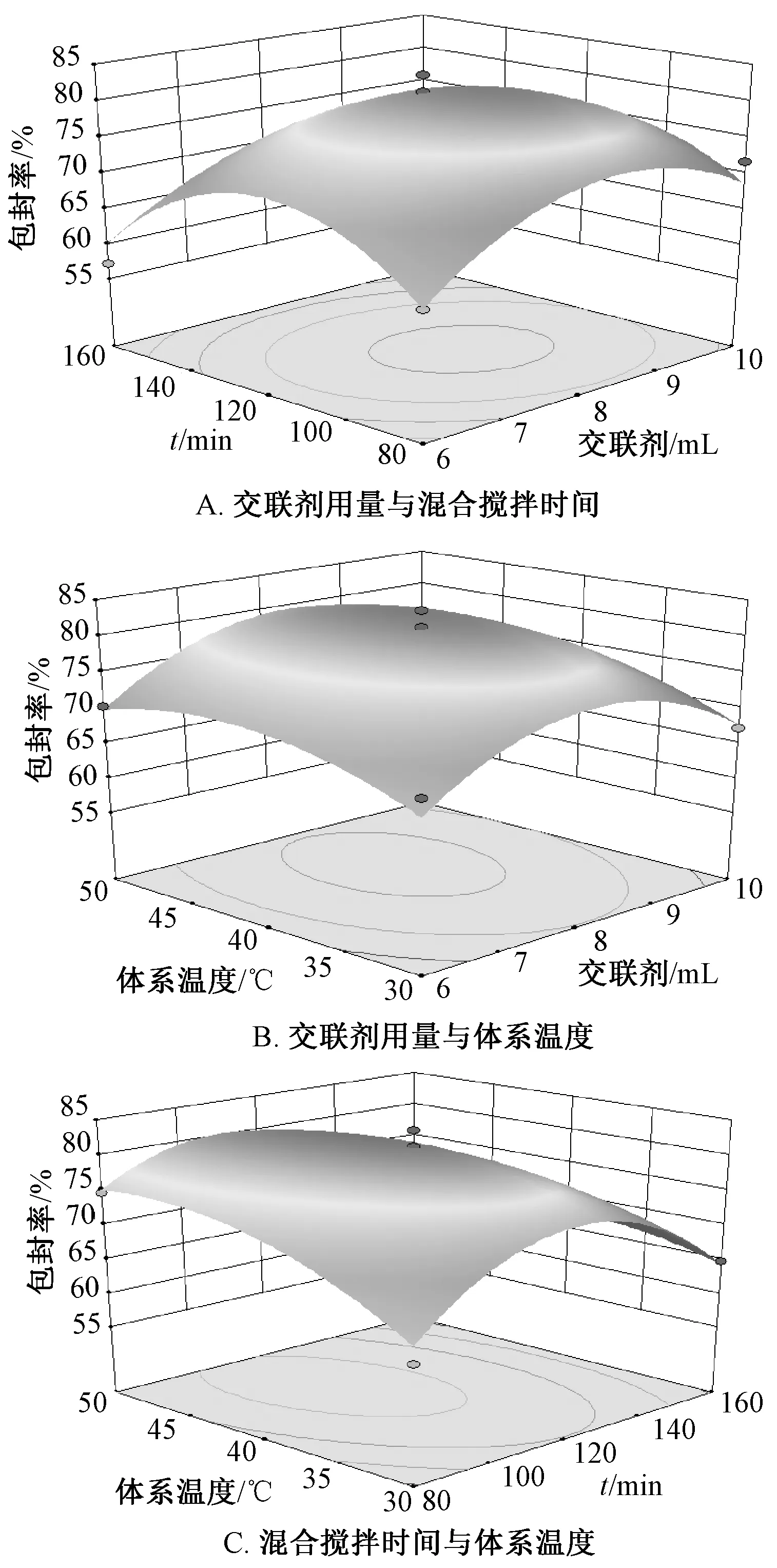
圖2 各因素響應面圖Fig.2 Respone surface plots for various factors
然后,按照優化工藝進行3批驗證試驗,結果見表3,可知包封率與預測值82.00%相當(相對誤差1.67%),表明工藝穩定可行,模型預測性良好。
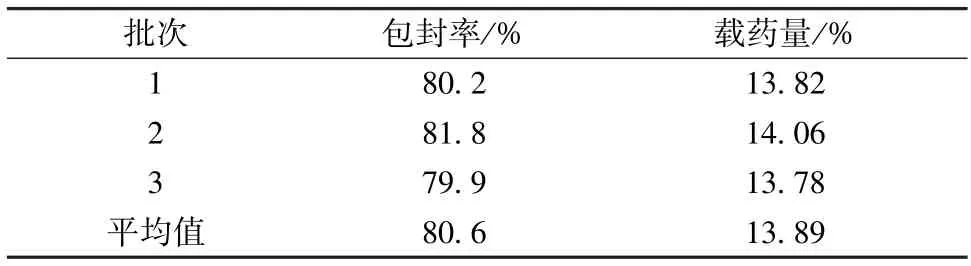
表3 驗證試驗結果(n=3)Tab.3 Results of verification tests(n=3)
2.5 微球表征
2.5.1 掃描電子顯微鏡(SEM)取適量微球均勻鋪在粘有導電膠的銅塊表面,噴金處理后進行觀察,結果見圖3。由圖可知,微球呈規則球形,表面較光滑,大部分粒徑在50 μm 左右。

圖3 微球SEM 圖Fig.3 SEM images for microspheres
2.5.2 傅立葉變換紅外光譜(FT-IR)KBr 將鹽酸小檗堿、殼聚糖微球、載藥微球、物理混合物分別壓片,在4 000~400 cm-1處測定紅外光譜,結果見圖4。由圖可知,鹽酸小檗堿1 623、1 503 cm-1附近是芳環骨架振動帶[14];殼聚糖微球除了殼聚糖特征峰(如3 400 cm-1的O-H 和N-H 伸縮振動峰,1 573 cm-1的 N-H 面內彎 曲振動 峰,1 046 cm-1的C-O 伸縮振動峰)[15]外,還有因戊二醛交聯而引入的C-H 伸縮振動峰2 941 cm-1(-CH3)和2 865 cm-1(-CH2);物理混合物可見明顯的鹽酸小檗堿、殼聚糖微球特征峰疊加;載藥微粒除了608 cm-1處有鹽酸小檗堿特征峰外,其余特征峰明顯減弱,表明微球實現了殼聚糖對鹽酸小檗堿的包覆,使后者較好地分散在微球內部。
2.5.3 X 射線衍射分析(XRD)圖5顯示,鹽酸小檗堿在18.29°、25.83°、28.64°,35.42°處有尖銳、狹窄的衍射峰,顯示出典型晶體結構[14];殼聚糖微球只在17°附近有1個較寬的特征峰;載藥微球除了26°附近有1個較弱的鹽酸小檗堿特征峰外,其余均被殼聚糖微球特征圖譜掩蓋;物理混合物可見明顯的鹽酸小檗堿、殼聚糖微球特征峰疊加,表明微球較好地包覆了鹽酸小檗堿。
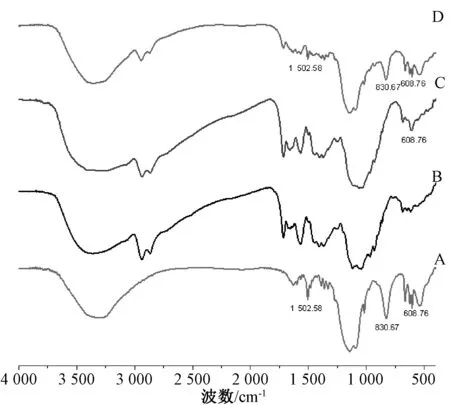
圖4 樣品FT-IR 圖Fig.4 FT-IR spectra for samples
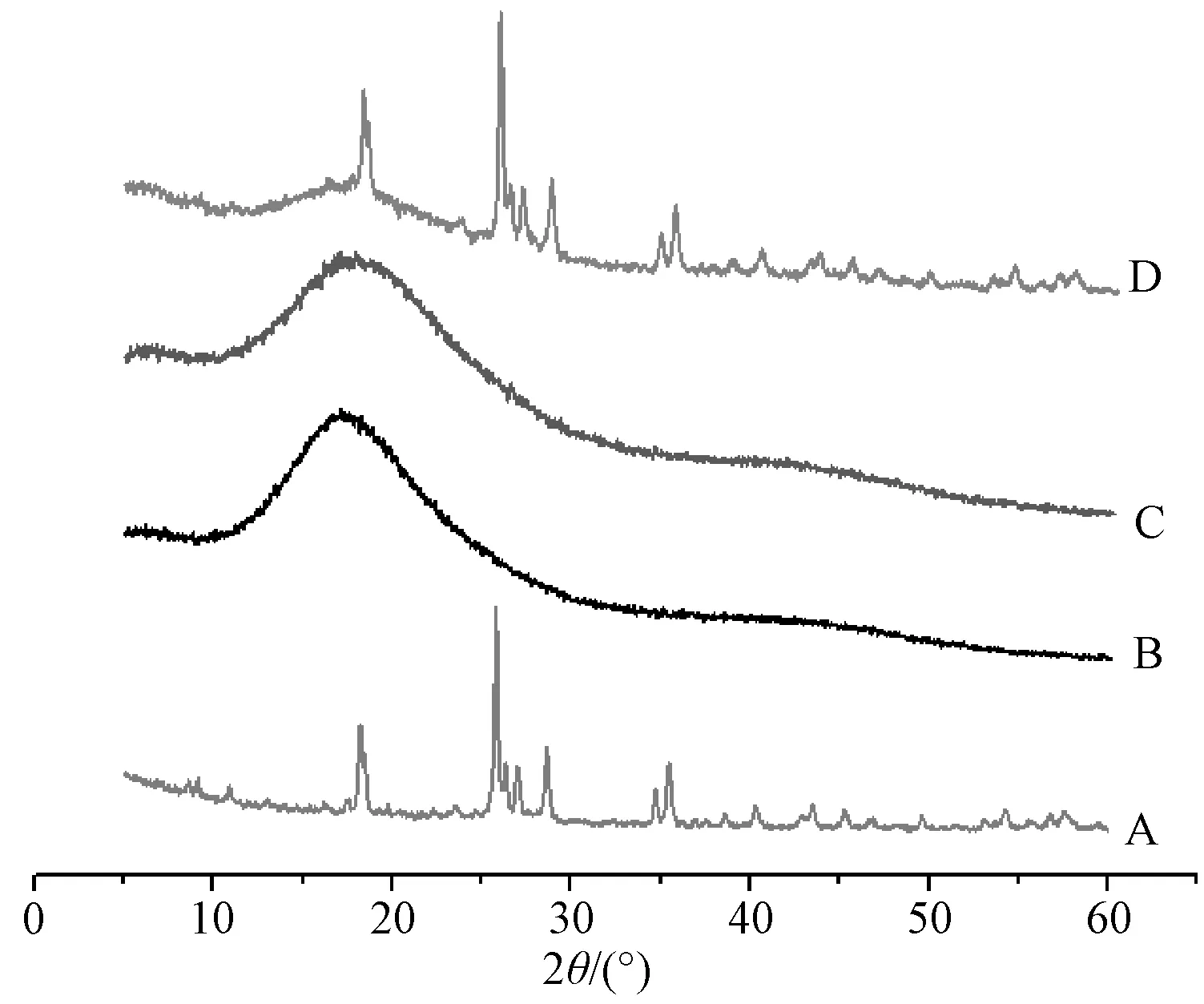
圖5 樣品XRD 圖Fig.5 XRD patterns for samples
2.6 體外釋放行為考察 以0.1 mol/L 鹽酸(pH=1.2)、磷酸鹽緩沖液(pH=7.4)為溶出介質,取相當于鹽酸小檗堿質量100 mg 的載藥微球,分散于5 mL 溶出介質后裝入透析袋內(截留分子量10 000 Da),置于95 mL 相同溶出介質中,(37± 0.5)℃恒溫水浴振蕩(100 r/min),于0.5、1、2、4、6、8、10、12、24 h 各取樣2 mL,并補充相同體積空白介質,0.22 μm 微孔濾膜過濾,在“2.2.1”項色譜條件下進樣測定,計算累積釋放率Q,公式為其中W為鹽酸小檗堿投入量,Cn為第n個取樣點溶液質量濃度,Ci為第n-1個取樣點溶液質量濃度,V為溶出介質總體積,Vi為取樣體積。結果見圖6。
由圖可知,微球在2種釋放介質中顯示出良好的緩釋性能,其中在0.1 mol/L 鹽酸(pH=1.2)中釋放更慢,24 h 內累積釋放率僅為77.8%,而在磷酸鹽緩沖液(pH=7.4)中達85.7%。同時,均未出現明顯突釋現象,0.5 h 內釋放率分別為4.8%、5.3%。
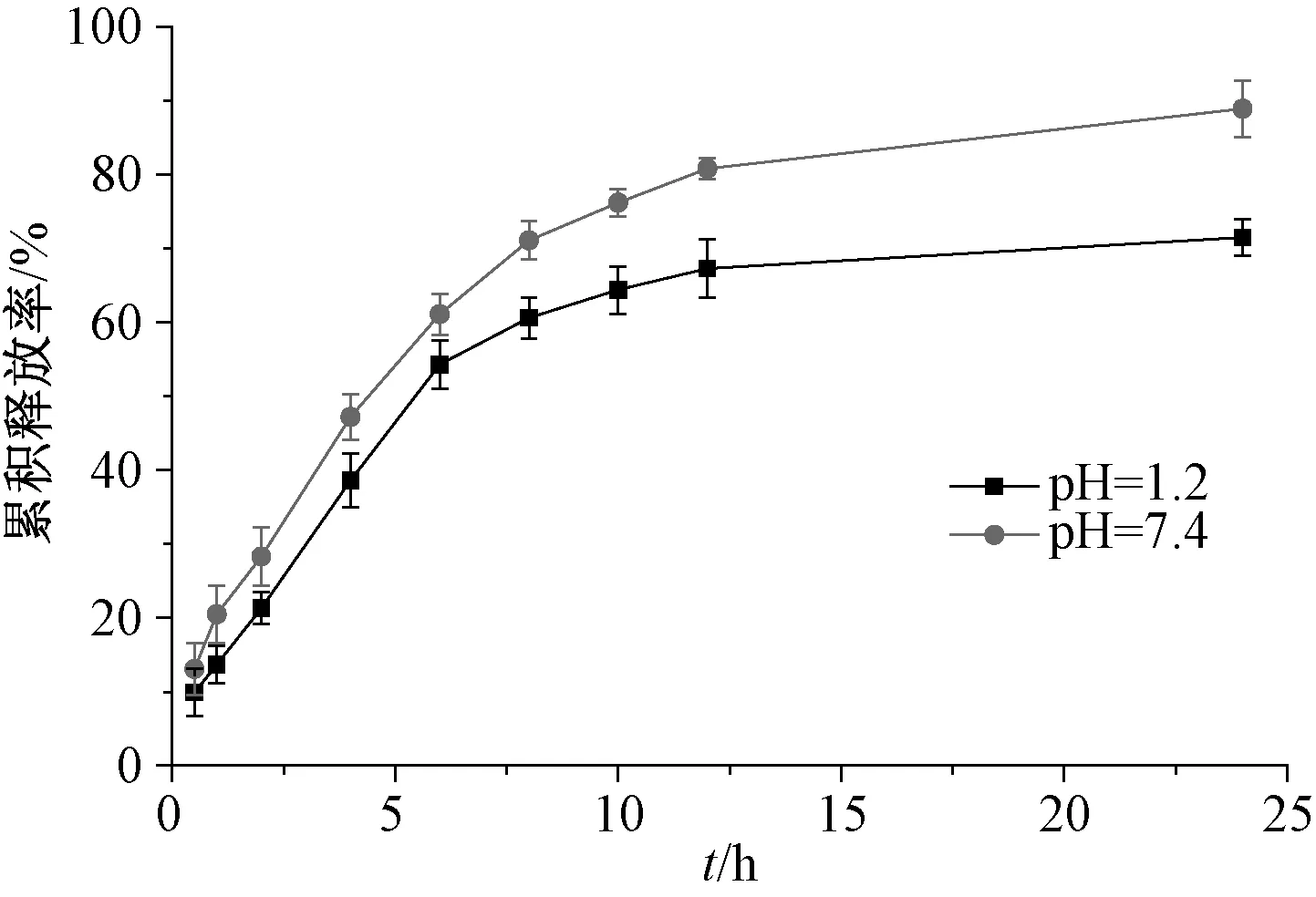
圖6 微球體外釋藥曲線Fig.6 In vitro drug release curves for microspheres
2.7 釋放動力學模型擬合 將微球體外釋放數據分別以零級、一級、Higuchi、Ritger-Peppas 模型進行擬合[16],結果見表4。由表可知,微球在2種釋放介質中均與Peppas 模型擬合度最高,n分別為0.789 2、0.778 9,屬于non-Fickian 擴散[17]。
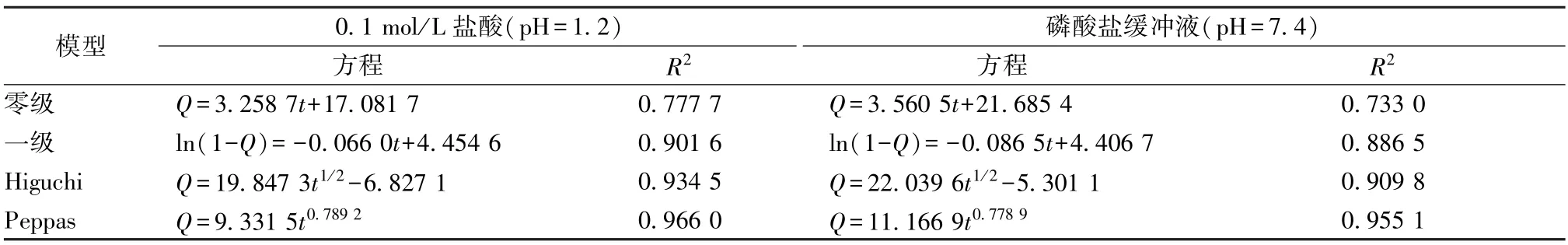
表4 微球模型擬合結果Tab.4 Results of model fitting of microspheres
3 討論
中藥單體活性成分的開發與利用是中醫藥走向現代化、國際化的重要基礎,近幾十年來青蒿素、小檗堿、穿心蓮內酯、川芎嗪等一大批中藥活性單體成分已成為臨床一線用藥,并受到國際學界高度關注[18]。然而,穩定性差、溶解度低等問題一直困擾著大部分中藥單體活性成分的開發利用,如丹參酮ⅡA 在水中的溶解度僅為2.8 ng/mL,半衰期僅為1~2 h[19],雖然納米粒、脂質體、微球等現代制劑技術可在一定程度上克服這些困擾[20],但穩定性、溶解度問題依然突出。
生物堿是一類含氮的堿性有機化合物,是中藥單體活性成分的重要組成部分,小檗堿、阿托品、茶堿、毛果蕓香堿等已廣泛應用與臨床[21]。但大多數生物堿幾乎不溶或難溶于水,導致其很難與親水性基質的水溶液共混制備藥物載體,只能以毒性相對較高的疏水性基質或價格較高的兩親性基質作為載體。
殼聚糖是一種廉價易得、性能優越的親水性藥物緩控釋載體基質材料,其分子中含有游離氨基,在酸性溶液中易成鹽,呈陽離子性質,在酸性環境中的溶解度高于在中性環境中[22],與生物堿的溶解特性相似,常用稀鹽酸或稀醋酸作為溶劑,其酸性溶液可促進生物堿溶解,有利于后者與其共混制備各類藥物載體。SPG 膜乳化技術制備殼聚糖微球技術成熟,操作簡便,制備條件溫和,無需高溫高壓,不會破壞藥物成分,而且載藥量高,易于規模化生產。
本實驗發現,殼聚糖/鹽酸小檗堿緩釋微球的最優制備工藝為交聯劑用量8 mL,混合攪拌時間112 min,體系溫度44 ℃,包封率80.6%,載藥量13.89%;微球在體外模擬胃液(pH=1.2)、腸液(pH=7.4)中都具有良好的緩釋性能,持續釋藥時間超過24 h,釋放均符合Peppas 模型和non-Fickian 擴散。由此可知,該微球可提高生物堿類成分在體液環境中的穩定性,并能減緩血藥濃度波動,降低給藥頻率,提高生物利用度,為相關緩控釋制劑研發提供實驗依據。
猜你喜歡
河北科技師范學院學報(2022年2期)2022-08-26 08:55:40
河北科技師范學院學報(2021年1期)2021-05-10 03:34:20
中成藥(2017年12期)2018-01-19 02:06:57
電源技術(2017年1期)2017-03-20 13:37:59
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
天然產物研究與開發(2016年1期)2016-06-05 10:29:25
食品界(2016年4期)2016-02-27 07:36:46
中國果菜(2015年2期)2015-03-11 20:01:01
應用化工(2014年7期)2014-08-09 09:20:21
應用技術學報(2014年4期)2014-02-28 14:52:40
