柴胡安心膠囊平行四波長高效液相色譜指紋圖譜研究*
2019-11-01 03:00:38焦麗亞張振杰王玲嬌楊繼章孫國祥周春華
中國藥業 2019年21期
關鍵詞:質量
于 靜,焦麗亞,張振杰,王玲嬌△,楊繼章,孫國祥 ,周春華
(1.河北醫科大學第一醫院,河北 石家莊 050031;2.沈陽藥科大學藥學院,遼寧 沈陽 110016)
柴胡安心膠囊為河北醫科大學第一醫院的醫院制劑(批準文號:冀藥制字Z20060013),由柴胡、葛根、白芍、桂枝、夏枯草等14味藥材組方,具有疏肝理氣、鎮靜安神、調和營衛之功效,主治焦慮抑郁和心臟神經官能癥[1]。有報道采用高效液相色譜(HPLC)法測定處方中單味藥材代表成分和建立單味藥材的指紋圖譜[2-4],柴胡安心膠囊(CHANCs)的質量標準目前只有顯微鑒別和薄層鑒別[5],尚未見關于其指紋圖譜研究方面的報道。單波長指紋圖譜鑒定的中藥質量具有隨機性和偶然性,多波長指紋圖譜可充分反映復方制劑在不同特征波長下的多維信息,更加準確地區分具有多個化學成分的復方制劑的質量等級[6-8]。本研究中建立了CHANCs的平行四波長指紋圖譜,采用系統指紋定量法鑒定其整體質量,為其質量標準的建立提供參考。
1 儀器與試藥
1.1 儀器
1200 Series型高效液相色譜儀(美國Agilent科技有限公司);Sartorius BS 110S型電子分析天平(北京賽多利斯科學儀器有限公司);KQ-400KDE型高功率數控超聲波清洗器(昆山市超聲儀器有限公司);微孔濾膜(0.45 μm,上海興亞凈化材料廠)。
1.2 試藥
沒食子酸對照品(批號為17062510),大豆苷對照品(批號為 17060913),肉桂酸對照品(批號為20171102),葛根素對照品(批號為 17072302),綠原酸對照品(批號為17081021),規格均為每瓶20 mg,均購于上海士鋒生物科技有限公司;蘆丁對照品(批號為20130809),槲皮素對照品(批號為 20121205),規格均為每瓶20mg,均購于美侖生物科技有限公司;柴胡安心膠囊(規格為每粒 0.5 g,批號分別為 150601,150518,150419,150407,080115,080302,080403,20180725,編號分別為S1~S8,河北醫科大學第一醫院)。
2 方法與結果
2.1 色譜條件與系統適用性試驗
色譜柱:DiamonsilC18柱(250mm ×4.6mm,5μm);流動相:0.5% 乙酸水(A)- 甲醇(B),梯度洗脫,0~40 min時5% ~25%B,40~60 min時25% ~60%B,60~80 min時 100%B;檢測波長:254,275,290,330 nm;流速:1 mL /min;進樣量:10 μL;柱溫:30 ℃。
樣品(批號為S2)制備供試品溶液并取混合對照品溶液,按擬訂色譜條件分別進樣,記錄275 nm波長下的色譜圖,詳見圖1。葛根素峰與相鄰峰分離較好,峰面積較大,理論板數不低于300 000,故選其作為參照峰。2 h空白溶液和供試品溶液色譜圖表明,檢測系統無其他峰干擾,組分在80 min內出峰完全。

圖1 高效液相色譜圖
2.2 溶液制備
稱取葛根素對照品30 mg,精密稱定,置10 mL容量瓶,用甲醇定容至刻度,搖勻,得對照品溶液。分別稱取大豆苷、沒食子酸、綠原酸、肉桂酸、蘆丁、槲皮素對照品(17,7,21,12,21,18 mg),用甲醇定容于 25 mL 容量瓶中,作為對照品貯備液。
稱取同一批樣品(批號為S2)內容物0.5 g,精密稱定,加入約50 mL 50%乙醇回流1.5 h,過濾,殘渣中加入50 mL 50%乙醇繼續回流1.5 h,重復2次,合并3次濾液,減壓濃縮至約15 mL,加50%乙醇定容至25 mL,搖勻,濾過,取續濾液,即得供試品溶液。
2.3 指紋圖譜檢測波長確定
處方中的柴胡藥效成分柴胡皂苷類吸收波長約在230 nm,白芍的芍藥苷最大吸收波長約為254 nm,葛根活性成分葛根素的最大吸收波長為280 nm,夏枯草有效成分迷迭香酸的最大吸收波長約為320 nm[9-12]。為使該復方制劑中的主要化學成分均能顯現最大紫外吸光度,盡可能地反映組方中代表成分的綜合特點,本研究中利用DAD檢測器掃描190~400 nm波長信號,依據具有藥效作用的藥材化學成分的紫外吸收光譜特征,最終選取了 4 個特征波長(254,275,290,330 nm)。
2.4 方法學考察
精密度試驗:精密吸取供試品溶液[取樣品(批號為S2)依法制備]10 μL,連續進樣 6 次,分別記錄 4 個檢測波長下的色譜圖。結果以葛根素峰的保留時間和峰面積為參照,四波長各指紋峰相對保留時間的RSD均小于1.00%,相對峰面積的RSD均小于 3.03% (n=6),表明儀器精密度良好。
穩定性試驗:精密吸取供試品溶液[取樣品(批號為S2)依法制備]分別于 0,4,8,12,24 h 時進樣測定,記錄4個檢測波長下的色譜圖。結果以葛根素峰的保留時間和峰面積為參照,四波長各指紋峰相對保留時間的RSD均小于 1.00%,相對峰面積的RSD均小于3.00%(n=6),表明供試品溶液在24 h內基本穩定。
重復性試驗:取樣品(批號為S2),平行制備供試品溶液6份,精密吸取10 μL進樣測定,分別記錄4個檢測波長下的色譜圖。結果以葛根素峰的保留時間和峰面積為參照,四波長各指紋峰相對保留時間的RSD均小于1.00%,相對峰面積的RSD均小于2.94% (n=6),表明方法重復性良好。
2.5 CHANCs四波長指紋圖譜分析
平行四波長指紋圖譜的建立:將8個批次樣品的供試品溶液分別進樣測定,記錄色譜圖。以葛根素峰為參照物峰,按峰出現率100.00% 計,確定各檢測波長下的共有指紋峰,分別是 63 個(254 nm)、65 個(275 nm)、53個(290 nm)、27個(330 nm)。將 8批樣品指紋圖譜信號導入“中藥色譜指紋圖譜超信息特征數字化評價系統4.0”軟件,按平均值法生成對照指紋圖譜(RFP),對照指紋圖譜和各個批次樣品HPLC指紋圖譜見圖2。
系統定量法綜合評價CHANCs平行四波長指紋圖譜質量:以生成的各個波長的對照指紋圖譜為標準,依次計算8批樣品的宏定性相似度(Sm)、宏定量相似度(Pm)、指紋均化性變異系數(α),詳見表 1。用均值法整合平行四波長HPLC指紋圖譜,計算Sm,Pm,α,用6級系統指紋定量法評價的質量結果見表2。結果顯示,8個批次樣品的Sm值均大于0.95,但 α值均小于0.15,在一定程度上表明不同批次樣品的平行指紋圖譜在化學成分數量和分布比例上有一定差異;Pm值除S1,S3,S7外,其余批次樣品均大于95%,表明不同批次樣品的宏定量相似度無顯著差異。最終評價的質量等級為S7為Ⅲ級,S1和S3為Ⅱ級,其余批次均為Ⅰ級。通常規定質量等級為Ⅴ級及以上時說明樣品質量合格,因此本研究中測定的8 批CHANCs 樣品均為合格品。

圖2 四波長下8批柴胡安心膠囊的高效液相色譜指紋圖譜和其對照指紋圖譜
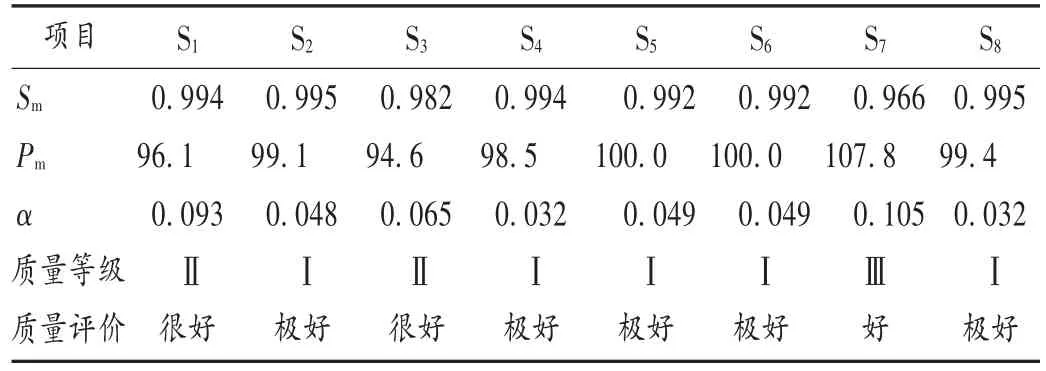
表1 系統指紋定量法評價柴胡安心膠囊質量結果

表2 柴胡安心膠囊平行指紋圖譜與各波長指紋圖譜結果
四波長下CHANCs的質量評價結果比較:將不同批次的樣品各個波長下分別建立對照指紋圖譜,采用系統指紋定量法評價其質量。結果見表3。可見,各樣品Sm和α相對偏差差異較小,表明指紋信號相似度、均化性與對照指紋圖譜差異較小。單波長的質量級別的劃分主要由Pm決定。不同評價方法的Pm值比較結果見圖3。4個特征波長和平行譜的質量等級比較結果見圖4,可見,同一樣品在不同波長下評價結果提示了個別批次樣品的質量差異很大,均值法整合的平行譜的質量等級和254 nm和330 nm波長下的質量等級結果相近,卻又有所不同,平行譜綜合了單個波長評價CHANCs質量的不足,使評價結果更接近真實結果。
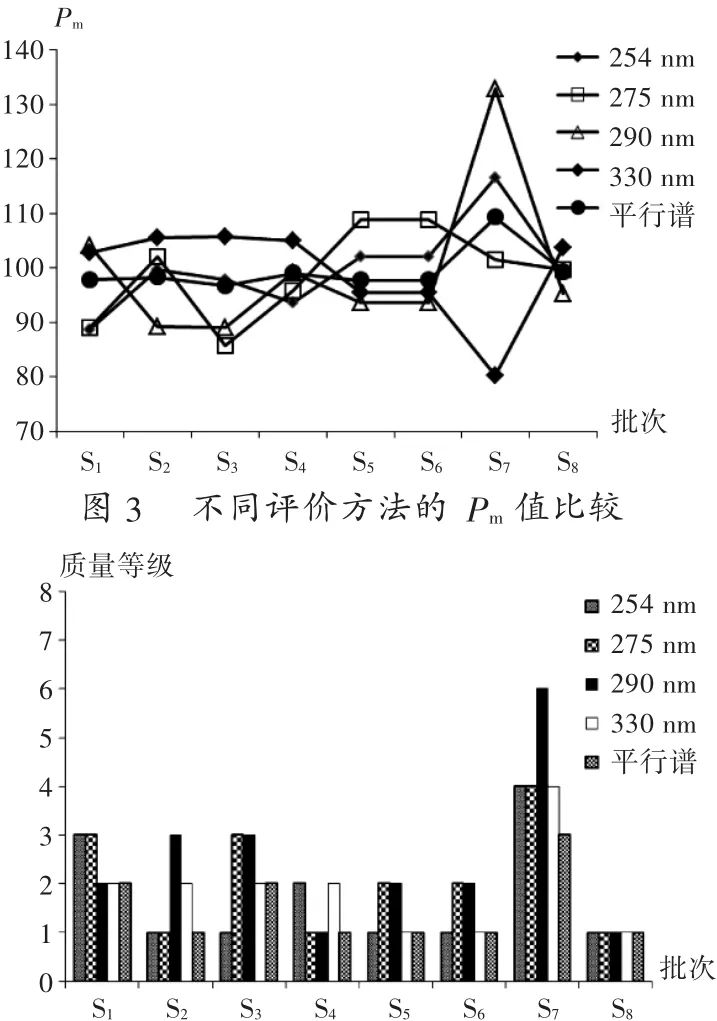
圖4 不同評價方法得到的樣品質量等級比較
3 討論
3.1 色譜條件選擇
柴胡安心膠囊主要藥材的成分如沒食子酸、大豆苷、肉桂酸等極性差異較大,故首選梯度洗脫。試驗過程中,發現色譜圖中4個主成分大峰難以分開,多次更換流動相比例和色譜柱均達不到基線分離,最后通過逐一單位藥材按供試品溶液方法處理進樣比對已知葛根中相關成分極性相差較小導致。故參考《中國藥典》的方法,選擇5% ~25%甲醇在40 min內緩慢升高梯度,使其相關峰得到分離。選擇流動相時,發現沒食子酸和大豆苷的色譜圖拖尾嚴重,選擇了不同體積分數的醋酸溶液,發現隨著醋酸比例的升高,上述成分的峰形逐漸對稱,進而達到測定要求。
3.2 平行指紋圖譜技術的均值法評價
利用任意的單個波長均不能完整地表達制劑中不同種類化學成分的全部指紋信息,因此選擇單味藥材中具有藥效作用的主要成分下的特征波長,對不同波長下色譜圖進一步整合,得到復方制劑的多維信息,才能最終鑒別其真實質量。本試驗中分別采用獨立權重法、均值法和投影參數法、信息熵權重法[13-14]整合各批次樣品在4個特征波長下的定性、定量指紋圖譜信息,以系統指紋定量法評價質量,發現8個批次的樣品質量等級基本一致。結合均值法簡單便捷的特點,最終選擇其作為整合指紋整體信息的方法。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
