不同配比青黛和雄黃對大鼠體內青黛指標性成分藥代動力學研究
2019-10-28 01:31:14吳青青汪電雷黃和平
安徽科技學院學報 2019年4期
關鍵詞:血漿
郭 艷, 吳青青, 汪電雷, 黃和平, 俞 娟
(安徽中醫藥大學 藥學院,安徽 合肥 230012)
青黃散是由青黛和雄黃兩味中藥組成,以青黛為君藥,具有涼血解毒、散瘀消積之功效,早在《世醫得效方》和《奇效良方》中就有記載,屬砷復合制劑[1]。方中青黛味咸、性寒,入肝經,能清熱解毒,涼血消斑,瀉火定驚,宜沖服或入丸散,其主要化學成分為靛藍、靛玉紅和色胺酮,具有抗菌消炎[2]、鎮痛[3]、保肝[4]、抗腫瘤[5]、免疫調節等作用[6-7]。靛玉紅是青黛抗白血病的主要藥效成分,對慢性粒細胞白血病具有明顯抑制作用[8]。雄黃辛溫有毒,可解百毒,消積聚,化腹中瘀血[9-10],其主要成分二硫化二砷(As2S2)可抑制腫瘤細胞增殖,降低端粒酶活性,以誘導細胞凋亡[11-14]。雄黃外敷具有消炎、抗病毒等作用[15],內服具有抗風濕、抗腫瘤的作用,常用于急性髓系白血病在內的多種惡性血液病的治療[16]。雄黃遇熱易轉化為有毒的As2O3,通過“水飛法”炮制雄黃以去除可溶性的As2O3,降低其毒性[17-18]。現代藥理研究表明,青黛與雄黃伍用可顯著增強其誘導白血病細胞凋亡的作用,常被用于血液疾病如白血病、骨髓增生異常綜合征等的治療[19-21]。研究發現,青黃散中青黛和雄黃不同劑量配比對體內砷的變化規律,結果表明青黃散中青黛比例的增加能促進大鼠體內砷的吸收[22]。為進一步考察青黛與雄黃配比變化對大鼠體內青黛指標性成分的藥動學行為及青黛吸收的影響,本實驗固定方中青黛的劑量,通過改變雄黃的劑量,采用UPLC-MS/MS法測定給藥后血中青黛指標成分色胺酮、靛玉紅和靛藍的濃度并比較其藥動學參數,深入探討其在體內的相互作用關系,為其在臨床應用提供理論依據。
1 材料與方法
1.1 實驗儀器
Agilent 1290超高效液相色譜儀(美國Agilent公司);Triple Quad TM 4500 質譜儀(美國AB SCIEX公司);AS5150A型超聲清洗儀(Autoscience公司);Dragonmed移液器(上海大龍醫療設備有限公司);BP211D型電子天平(德國Sartorius公司);Milli-Q超純水器(美國 Millipore公司)。
1.2 供試材料
青黛(批號:170907)和雄黃(批號:171023)均購于安徽省亳州中藥材市場,并經安徽中醫藥大學彭華勝教授鑒定為正品。標準品(純度均≥98%):靛玉紅(批號:110717)和靛藍(批號:67707224)均購于中國食品藥品檢定研究院;色胺酮(韶遠科技上海有限公司,批號:SY017117);卡馬西平(成都德斯特生物技術有限公司,批號DST171120-318);N,N-二甲基甲酰胺(色譜純,上海阿拉丁生化科技有限公司);乙腈(色譜純,合肥科申化工有限公司);水為超純水,其余試劑均為分析純。
1.3 試驗動物
雄性SD大鼠,體質量為180~220 g,由安徽醫科大學實驗動物中心提供,合格證號:SCXK(皖)2017-001,實驗前禁食12 h,可自由飲水。
1.4 試驗方法
1.4.1 色譜條件與質譜條件 Kinetex C18(100 mm×2.10 mm, 1.7 μm)色譜柱,流動相為乙腈-水(53∶47,V/V),流速為0.2 mL/min, 柱溫為30 ℃,進樣量為2 μL。采用電噴霧離子化(ESI)以多反應監測模式(MRM)進行正離子掃描。氣路參數:噴霧電壓(IS)為5.5 kV,離子化溫度(TEM)為550 ℃;碰撞氣(CAD)為10 psi、霧化氣(GS1)為18 psi、加熱氣(GS2)為55 psi。用于定量分析的離子對和去簇電壓(DP)及碰撞電壓(CE)分別為:m/z 249.2→130.0,60 eV,38.63 eV(色胺酮);m/z 263.0→235,77.22 eV,34.3 eV(靛玉紅);m/z 263.0→235,77.22 eV,34.3 eV (靛藍);m/z 237.2→194.1,104.04 eV,27.8 eV (卡馬西平)。
1.4.2 供試品的制備 稱取1.0 g雄黃于研缽中,加0.5 mL水研磨5 min至糊狀,再向研缽中加入40 mL水攪拌1 min,靜置4 min,取混懸液,下沉的粗粉繼續研磨,反復操作10次,合并混懸液,靜置10 h以上,取下沉物冷凍干燥[23-24]。稱取3份青黛粉末各10.0 g,按照190.0∶1、95.0∶1、47.5∶1的配比分別稱取上述冷凍干燥后的雄黃52.6、105.3、210.5 mg,然后均以18 mL 0.5% CMC-Na和2 mL乙醇制成溶液,灌胃給藥,給藥體積為10 mL/kg。
1.4.3 對照品及內標溶液的配制 稱取適量各對照品,分別用N,N-二甲基甲酰胺溶解,得到濃度均為100 μg/mL的各對照品儲備液。分別移取不同體積各儲備液于10 mL容量瓶中, 乙腈定容, 得色胺酮、靛玉紅和靛藍濃度分別為200、2 000和2 000 ng/mL的混合對照品儲備液。精密稱取卡馬西平1.00 mg于10 mL容量瓶,乙腈溶解并定容,得濃度為100 μg/mL的內標儲備液。取適量該儲備液,用乙腈稀釋成濃度為0.1 ng/mL的內標溶液,4 ℃下保存備用。
1.4.4 血漿樣品處理 取血漿90 μL置于1.5 mL EP管中,加入10 μL內標和750 μL乙酸乙酯,渦旋3 min,12 000 r/min離心10 min,取上清液,氮吹,加50 μL乙腈復溶,渦旋混勻,再次12 000 r/min離心10 min,取上清液。
1.5 藥代動力學實驗
取18只SD大鼠,隨機分為A、B、C三組,實驗前禁食12 h,可自由飲水。以青黛劑量為5 g/kg,雄黃劑量分別為0.026 g/kg(A組)、0.053 g/kg(B組)、0.105 g/kg(C組)的配比進行大鼠灌胃給藥,并于給藥前和給藥后5、15、30、60、120、240、360、480、600 min眼底靜脈叢取血0.3 mL,置于經肝素處理的EP管中,3 500 r/min離心10 min,分離出血漿,按“1.4.4”節下血漿樣品處理操作,進樣分析。
1.6 數據處理
運用DAS 2.0軟件進行藥動學參數的計算。采用SPSS 23.0軟件對數據進行分析,結果以“均值±標準差”表示,P<0.05表示差異顯著。
2 結果與分析
2.1 專屬性考察
分別取大鼠空白血漿、空白血漿加內標、空白血漿加內標和混合對照品溶液以及給藥6 h后的大鼠血漿樣品,按“1.4.4”節進樣分析。如圖1所示, 血漿中內源性物質對色胺酮、靛玉紅、靛藍和卡馬西平的測定無干擾。
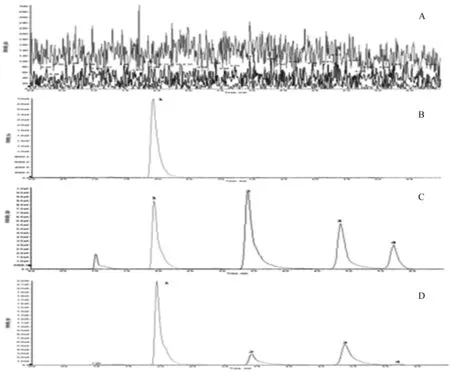
圖1 大鼠空白血漿(A)、空白血漿加內標(B)、空白血漿加內標和混合對照品溶液(C)以及青黛∶雄黃=95∶1配伍給藥6h后的大鼠血漿樣品(D)的MRM色譜圖,1.卡馬西平;2.色胺酮;3.靛藍;4.靛玉紅Fig.1 MRM chromatograms of rat blank plasma (A), blank plasma plus internal standard (B), blank plasma plus internal standard and mixed reference solution (C), as well as rat plasma samples after 6 h of administration (D, indigo naturalis∶realgar=95∶1), 1. Carbamazepine; 2. Tryptanthrin; 3. Indigo; 4. Indirubin
2.2 標準曲線的繪制
用乙腈依次稀釋混合對照品儲備液,得到色胺酮濃度為2,4,10,20,50,100,200 ng/mL,靛玉紅和靛藍濃度分別為20,40,100,200,500,1 000,2 000 ng/mL的混合對照品溶液。取大鼠空白血漿90 μL,分別加入10 μL一系列混合工作液和750 μL乙酸乙酯,按“1.4.4”節下進樣分析。以樣品濃度為橫坐標(x),以樣品與內標物峰面積比值為縱坐標(y)進行線性回歸,繪制標準曲線。得到方程:色胺酮:y=0.557x+0.327 4(r=0.996 1);靛玉紅:y=0.010 3x-0.000 6(r=0.998 2);靛藍:y=0.053 7x+0.229 6(r=0.997 3)。結果表明,色胺酮在0.2~20 ng/mL 濃度范圍內線性關系良好,靛玉紅和靛藍在2~200 ng/mL濃度范圍內線性關系均良好。
2.3 準確度考察
取大鼠空白血漿90 μL,加入一定量不同濃度的混合對照品溶液,分別配制成色胺酮、靛玉紅和靛藍定量下限(0.2,2,2 ng/mL)、低(0.4,4,4 ng/mL)、中(2,20,20 ng/mL)、高(10,100,100 ng/mL)濃度的質控樣品,每個濃度平行5份,重復3個分析批,按“1.4.4”節下進樣分析。結果如表1~3所示。
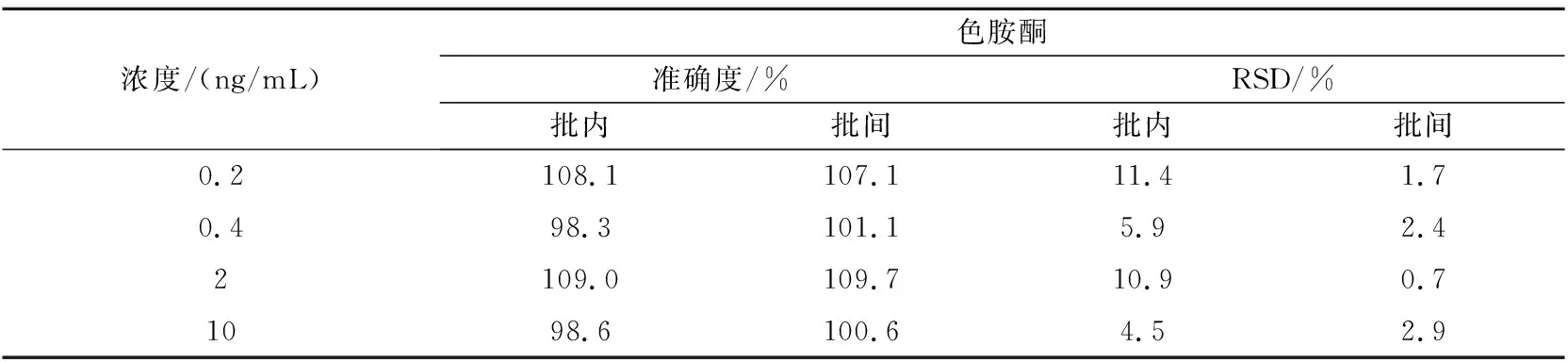
表1 色胺酮準確度測定(n=5)
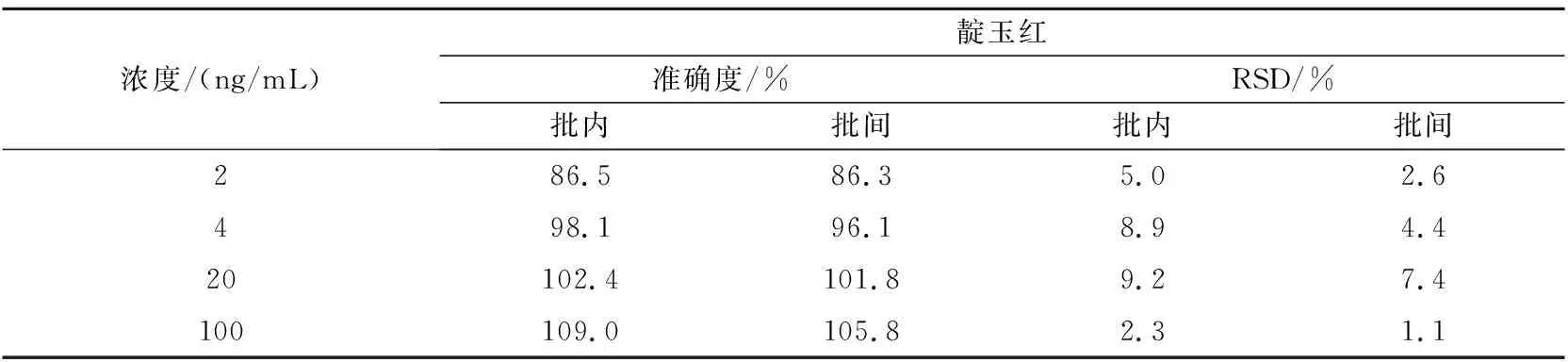
表2 靛玉紅準確度測定(n=5)
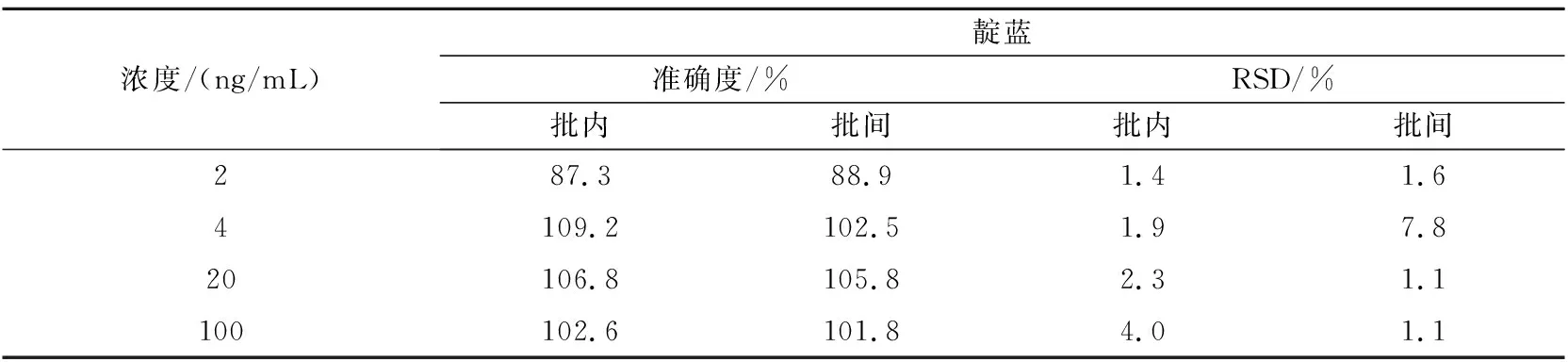
表3 靛藍準確度測定(n=5)
2.4 基質效應
以大鼠空白血漿經乙酸乙酯處理后的空白基質上清液和乙腈為溶劑,分別配制含基質和不含基質的色胺酮、靛玉紅和靛藍低、中、高濃度的質控樣品,每個濃度平行5份,進樣分析。每個分析物及內標的基質因子為基質存在下的峰面積(由乙酸乙酯處理后的空白血漿加入分析物和內標測得)與不含基質的相應峰面積(分析物和內標的純溶液)的比值。分析物與內標的基質因子的比值即為內標歸一化的基質因子。結果表明內標歸一化的基質因子的RSD均小于15%, 表明血漿基質對3種物質的測定影響較小。
2.5 提取回收率
以空白血漿90 μL,加入色胺酮、靛玉紅和靛藍低、中、高濃度的混合對照品溶液, 按血漿樣品處理方法處理后的測定峰面積為A1,空白血漿樣品按血漿樣品處理方法處理后加入色胺酮、靛玉紅和靛藍低、中、高濃度的混合對照品測定的峰面積為A2,提取回收率=A1/A2×100%。結果表明低、中、高濃度下3種物質的提取回收率均大于85%,符合生物樣品分析要求。
2.6 穩定性考察
配制色胺酮、靛玉紅和靛藍低、中、高濃度的質控樣品,每個濃度平行5份。樣品分別經反復凍融3次、室溫放置6 h以及4 ℃放置24 h后, 按“1.4.4”節下進樣分析。結果表明低、中、高濃度下3種物質的RE均小于15%,表明這3種物質在上述條件下均穩定。
2.7 青黛三種指標成分的血藥濃度-時間曲線及其主要藥代動力學參數
色胺酮、靛玉紅和靛藍的平均血藥濃度-時間曲線如圖2~4所示。采用DAS 2.0軟件進行分析,得到的主要藥動學參數結果見表4~6。
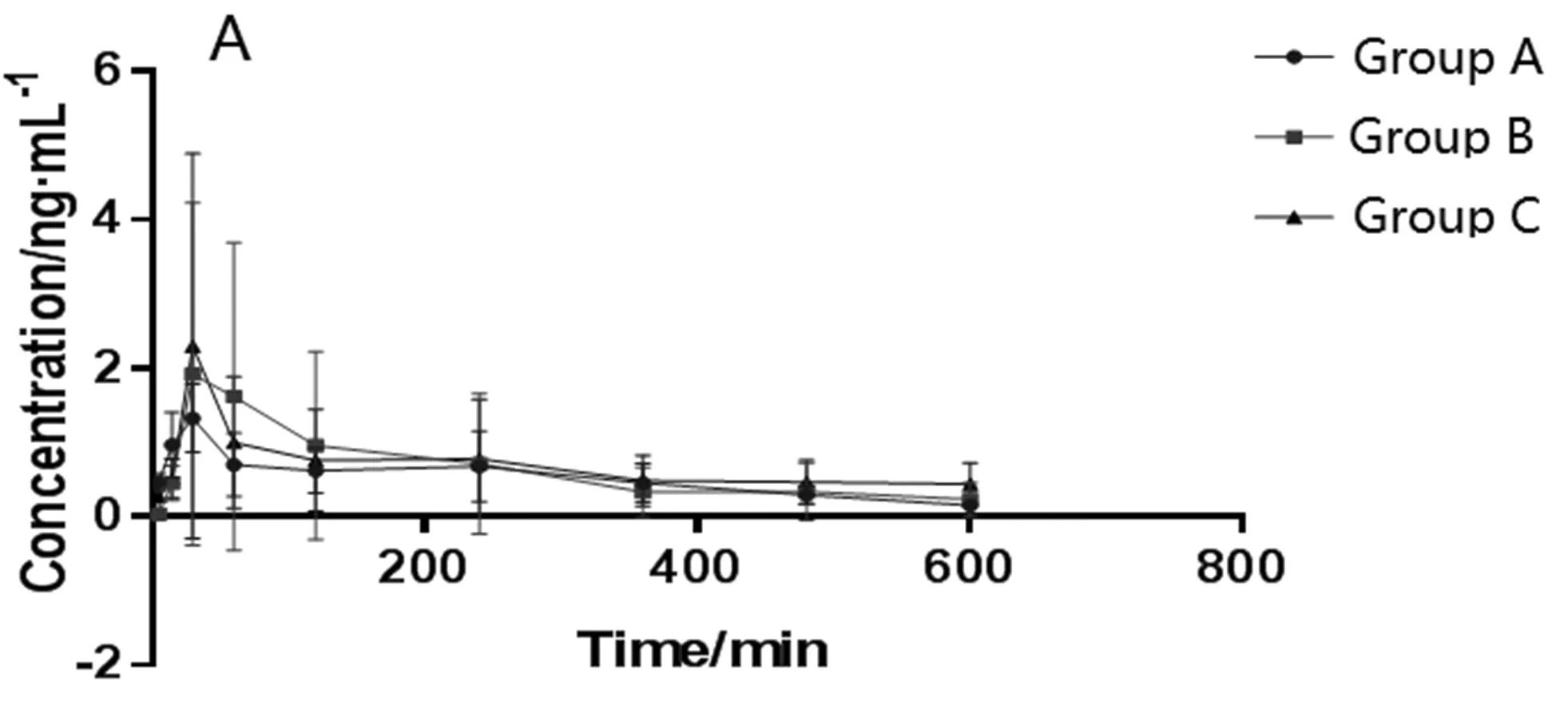
圖2 色胺酮平均血藥濃度-時間曲線(n=6)Fig.2 Mean plasma concentration-time curve of tryptanthrin
圖3 靛玉紅平均血藥濃度-時間曲線(n=6)Fig.3 Mean plasma concentration-time curve of indirubin
圖4 靛藍平均血藥濃度-時間曲線(n=6)Fig.4 Mean plasma concentration-time curve of indigo
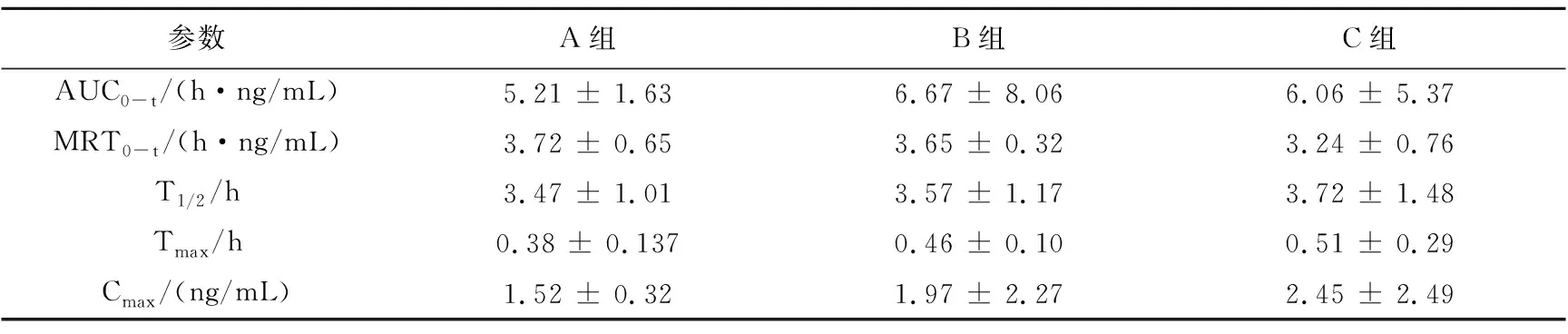
參數A組B組C組 AUC0-t/(h·ng/mL)5.21 ± 1.636.67 ± 8.066.06 ± 5.37 MRT0-t/(h·ng/mL)3.72 ± 0.653.65 ± 0.323.24 ± 0.76 T1/2/h3.47 ± 1.013.57 ± 1.173.72 ± 1.48 Tmax/h0.38 ± 0.1370.46 ± 0.100.51 ± 0.29 Cmax/(ng/mL)1.52 ± 0.321.97 ± 2.272.45 ± 2.49
表5 大鼠灌胃給藥后靛玉紅主要藥動學參數
注:C組與A組比較,*P< 0.05;C組與B組比較,#P< 0.05。下同。

表6 大鼠灌胃給藥后靛藍主要藥動學參數
3 結論與討論
前期研究比較了甲醇、乙腈和N,N-二甲基甲酰胺等溶劑超聲溶解效果,結果顯示色胺酮、靛玉紅及靛藍在N,N-二甲基甲酰胺中均能較好地溶解,故實驗選用N,N-二甲基甲酰胺溶解這三種物質。卡馬西平以及各對照品在相同色譜條件下出峰位置合適,且無組分峰影響,實驗選用卡馬西平為內標物。青黛難溶于水,實驗采用0.5%CMC-Na和少量乙醇溶解青黛粉末以制成混懸液灌胃給藥。關于生物樣品前處理,用乙酸乙酯萃取青黛中的靛玉紅,乙腈沉淀蛋白效果好,氮吹效率高[23]。本實驗還考察了甲醇-水、乙腈-水的不同比例流動相,結果表明以乙腈-水(53∶47,V/V)作為流動相時,待測物響應值較高,分離效果較好,因而實驗選用乙腈-水(53∶47,V/V)為流動相進行指標成分濃度的測定。
一般來說,中藥復方不同配比的選擇可直接影響其臨床療效與安全[24]。青黃散中隨著雄黃劑量的增大,其治療白血病的效果增加,但雄黃劑量過大會造成體內砷蓄積而引起一系列毒副作用[25]。青黃散中青黛與雄黃常用比例為7∶3、8∶2、9∶1,但可能由于儀器等實驗條件受限,前期預實驗結果發現,青黛在較低劑量時很難同時測定青黛中三種指標成分的體內濃度。本實驗在前人工作的基礎上,通過比例調整,分別以青黛與雄黃比例為190.0∶1、95.0∶1和47.5∶1進行大鼠灌胃給藥,測定給藥后血中青黛指標成分的濃度并比較其藥動學參數,探討不同配比的青黛與雄黃伍用對體內青黛吸收的影響。結果顯示,隨著雄黃劑量的增大,色胺酮的Cmax、AUC0-t和MRT均無顯著變化(P>0.05),說明雄黃劑量變化不影響色胺酮的吸收;靛藍的Cmax和AUC0-t均顯著增大(P<0.05),說明雄黃劑量增加能促進青黛中靛藍的吸收[22];靛玉紅的Cmax和AUC0-t均減小(P<0.05),可能是由于青黛中的靛玉紅與雄黃中的砷化物相互作用形成有機配合物發揮其抗腫瘤作用而使血中靛玉紅濃度降低,這可能是青黛與雄黃伍用治療白血病效果更優的一個原因[26]。綜上可知,不同配比的青黛與雄黃伍用時,青黛的體內藥動學行為發生改變,雄黃劑量增大能促進青黛吸收,但不是促進全部有效成分吸收。而對于青黛與雄黃伍用治療血液疾病的確切作用機制還有待進一步研究。
猜你喜歡
現代臨床醫學(2022年4期)2022-09-29 07:38:00
昆明醫科大學學報(2021年4期)2021-07-23 01:21:50
天津醫科大學學報(2019年6期)2019-08-13 07:04:34
云南醫藥(2019年3期)2019-07-25 07:25:14
現代檢驗醫學雜志(2016年5期)2016-08-20 03:16:56
海南醫學(2016年8期)2016-06-08 05:43:00
西南軍醫(2016年5期)2016-01-23 02:20:33
川北醫學院學報(2015年5期)2015-12-05 08:22:28
醫學研究雜志(2015年9期)2015-07-01 17:28:15
現代檢驗醫學雜志(2015年1期)2015-02-06 01:59:26
