多種遺傳學技術聯合運用對疑似Meckel綜合征家系進行遺傳學分析
2019-10-21 03:09:26張靚璠嚴愷王英黃莉
溫州醫科大學學報 2019年10期
張靚璠,嚴愷,王英,黃莉
(1.義烏市婦幼保健院 計劃生育服務中心,浙江 金華 322000;2.浙江大學醫學院附屬婦產科醫院生殖遺傳科,浙江 杭州 310000)
Meckel綜合征是一種致死性的常染色體隱性遺傳病,由Meckel等在1822年首次報道。典型臨床癥狀包括腎囊腫、內生殖器異常、軸后多指畸形、肝纖維化、無腦畸形、唇腭裂、枕葉腦膨出等。
該病在不同國家新生兒中的發病率不同,在英國約為1/140 000[1],在芬蘭約為1/9 000[2],在印度古吉拉特邦中發病率則高達1/1 304[3],而在我國目前僅見散發病例報道,尚無相關數據統計。該病預后差,胎兒常發生宮內死亡,出生的胎兒大多僅能存活數天至數周且沒有治療措施,因此臨床上與該疾病相關的研究較少。隨著基因測序技術的發展,基因檢測技術已成為一種發現新致病位點和進行遺傳病診斷的有效途徑,并在不斷地發展與完善。本研究中,我們對義烏市婦幼保健院收集到的1例疑似Meckel綜合征的引產胎兒組織及其家系成員進行了遺傳學檢測,利用全外顯子高通量測序技術及Sanger測序技術發現了CC2D2A基因上的2個致病性突變c.3964C>T和c.4567T>C。本研究豐富了Meckel綜合征的致病性突變數據庫,并為該家系遺傳咨詢、產前診斷及胚胎植入前診斷提供了遺傳學依據。
1 對象和方法
1.1 對象先證者母親,G5P2,既往體健,2007年行甲狀腺結節切除術,術后恢復尚可,無高血壓、糖尿病、心臟病等疾病史,無肝炎、結核等傳染病史,無輸血史,無明顯藥物、食物過敏史,無長期藥物使用史,無藥物成癮。第1胎足月自然分娩女嬰,健康;第2胎自愿人工流產;第3、第4胎,均為男胎,分別于妊娠13+和19+周時,B超顯示枕葉腦膨出和多囊腎,經慎重考慮終止妊娠;第5胎,足月自然分娩一健康女嬰。家系圖譜見圖1。
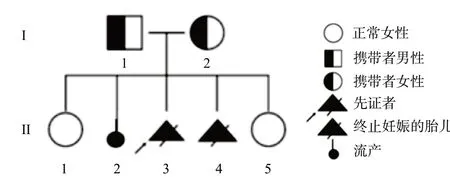
圖1 Meckel綜合征家系圖譜
1.2 方法根據先證者胎兒超聲結構異常的相關結果,我們對第3胎引產胎兒組織及其父母、姐妹進行了遺傳學檢測。該研究項目通過了本院醫學倫理委員會的審核批準,并取得了先證者父母簽署的知情同意書。取先證者胎兒腓腸肌組織,用0.9%氯化鈉溶液漂洗排除母血污染,使用基因組雜交芯片(comparative genomic hybridization,CGH)和全外顯子組測序(whole exome sequencing,WES)技術對先證者染色體組及外顯子組進行檢測;分別取先證者父母及其姐妹的EDTA抗凝外周血5 mL,針對先證者發現的致病位點,通過Sanger測序對家系進行驗證。
1.2.1 CGH檢測:按照Qiagen試劑盒標準流程提取血液中基因組DNA。染色體芯片采用Agilent Sure-Print G3 Custom CGH+SNP Microarray(4×180 K),操作步驟簡述如下:用限制性內切酶酶切對照及樣本基因組,以Cy3-dUTP和Cy5-dUTP分別標記對照和樣本基因組DNA,純化后測定標記效率及DNA量,將已標記的對照及樣本基因組DNA混合,位于芯片上雜交,洗滌芯片后選擇合適的掃描通道在Agilent掃描儀上掃描,掃描結果經計算機轉換,得到每個位點的相對信號強度,進行作圖與分析。
1.2.2 WES檢測:超聲將胎兒基因組DNA打斷,獲得200~250 bp大小的DNA片段,純化后,對DNA片段進行末端修復、添加單堿基A以及測序接頭,篩選出片段適中的DNA片段構成原始文庫,PCR擴增后完成DNA建庫。使用SeqCap EZ Human Exome Probes v3試劑盒,將文庫與探針庫進行雜交,捕獲基因的外顯子,洗滌后將外顯子DNA富集,再次PCR擴增,純化后得到測序文庫。最后利用高通量測序儀HiseqX TEH連續雙向測序(平均測序深度不低于100×),用FASTX-Toolkit軟件讀出原始測序數據。
1.2.3 序列分析:首先對原始測序數據進行質量評估,去除低質量及被污染的測序數據,隨后用Burrows Wheeler Aligner(BWA)軟件進行序列比對(比對參照hg19),同時評價序列捕獲效果,用GATK軟件和VarScan軟件分別進行單核苷酸變異(SNP)和插入缺失(InDel)查詢,得到目標區域堿基多態性結果,發現可疑的變異位點。在可疑的變異位點中去除內含子區變異,并在EXAC數據庫(http://exac.broadinstitute.org)、1 000 human genome數據庫(http://www.internationalgenome.org/data)中過濾頻率大于1%的變異位點,添加HGMD數據庫(http://www.hgmd.cf.ac.uk/ac)、OMIM數據庫(http://www.omim.org)的相關注釋信息,最后應用SIFT(http://sift.jcvi.org)、PolyPhen-2(http://genetics.bwh.harvard.edu/pph2)、Mutation Taster(http://www.mutationtaster.org)軟件對基因突變位點的致病性進行分析,使用ExPASy(https://www.expasy.org)軟件預測突變位點對蛋白結構和功能的影響,以美國醫學遺傳學與基因組學會(American College of Medical Genetics and Genomics,ACMG)發布的《ACMG遺傳變異分類標準與指南》(2015)對發現的變異位點進行分類。
1.2.4 Sanger測序驗證:采用ABI 3730測序儀驗證高通量測序發現致病性突變,根據突變所在位置,在其上、下游設計相應的特異性引物。通過電泳確認PCR反應后,對片段進行Sanger測序,測序結果用Chromas軟件讀取,并與NCBI(https://www.ncbi.nlm.nih.gov)中的基因標準序列進行比對分析,同時驗證高通量測序結果與Sanger測序結果一致性。
2 結果
2.1 CGH檢測本研究首先利用CGH技術檢測先證者胎兒是否存在致病性拷貝數變異、雜合性缺失或單親二倍體。結果顯示先證者胎兒染色體不存在上述3種致病性染色體異常,見圖2。
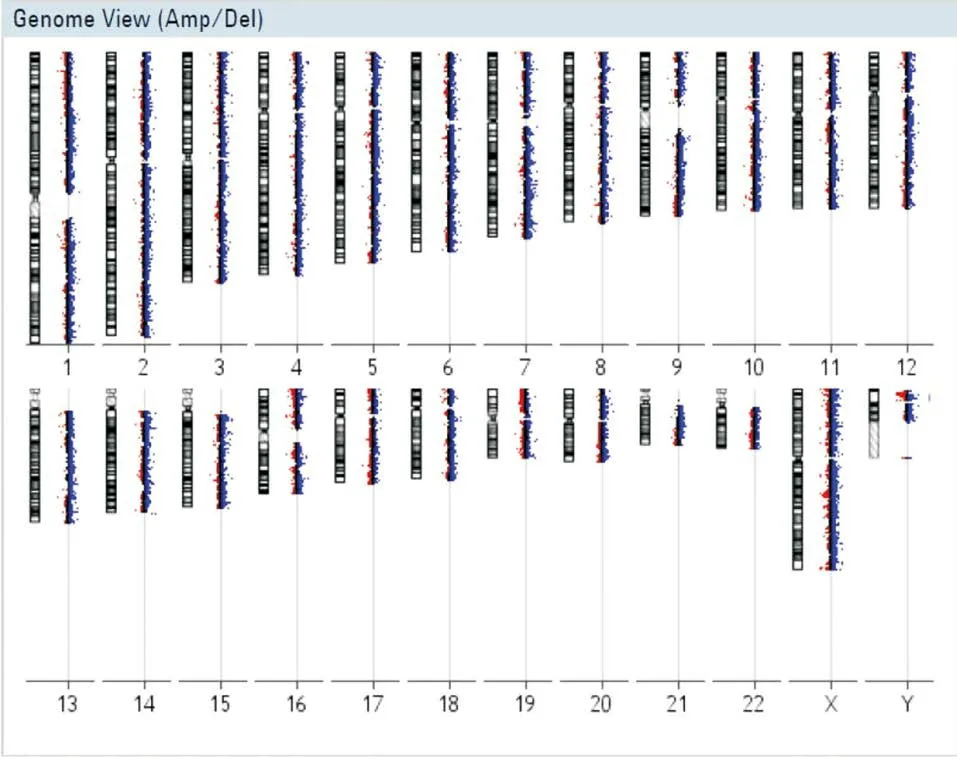
圖2 先證者染色體微陣列芯片檢測結果
2.2 WES檢測采用全外顯子測序對先證者胎兒組織的基因組DNA進行檢測。檢測結果顯示,先證者存在CC2D2A(NM_001080522):c.3964C>T(p.Q1322X)和c.4567T>C(p.C1523R)復合雜合突變,見表1。
2.3 突變位點的功能預測c.3964C>T(p.Q1322X)為無義突變,該突變可將編碼CC2D2A蛋白中第1 322號位置谷氨酰胺的密碼子(CAG)變為終止密碼子(TAG),從而導致編碼蛋白提前終止,產生截短蛋白或蛋白被降解;該突變在人群中發生頻率極低(ExAC、HGMD、ClinVar數據庫均未見收錄);Polyphen、SIFT、MutationTaster軟件預測可能影響蛋白功能尚未見此變異在患者中檢出的文獻報道。根據《ACMG遺傳變異分類標準與指南》(2015)將其評級為致病(PVS1+2*PP)。
c.4567T>C(p.C1523R)的變異描述和上面相似,SIFT、PolyPhen-2、Mutation Taster軟件對該變異的致病性進行分析,結果均提示該突變有害;ExAC、HGMD數據庫未見收錄。根據《ACMG遺傳變異分類標準與指南》(2015)將其評級為可能致病(2*PM+2*PP)。
2.4 Sanger 測序驗證在發現先證者中存在CC2D2A基因(NM_001080522)的c.3964C>T(p.Q1322X)和c.4567T>C(p.C1523R)突變后,在先證者的父母及其姐妹中檢測是否存在上述突變。Sanger測序結果顯示,先證者的母親攜帶CC2D2A基因c.3964C>T(p.Q1322X)突變的雜合變異。先證者父親攜帶c.4567T>C(p.C1523R)雜合變異。見圖3。因此先證者所攜突變分別遺傳自父母雙方。

表1 先證者CC2D2A基因變異分析結果
3 討論
Meckel綜合征為一種罕見的致死性常染色體隱性遺傳病,于1969年Opitz和Howe首次描述了Meckel綜合征的臨床特征[4]。該綜合征的表型包括囊性腎發育不良、枕部腦膨出或其他中樞神經系統異常以及多指(趾)畸形,此3 種表型也是確診Meckel綜合征的主要標準,臨床上,孕11~14周常規超聲檢查發現2種及以上典型的Meckel綜合征表型時,即可明確診斷疾病。本研究中,先證者胎兒表現出了腦膨出和多囊腎異常,符合Meckel綜合征的臨床診斷標準,可診斷為Meckel綜合征。
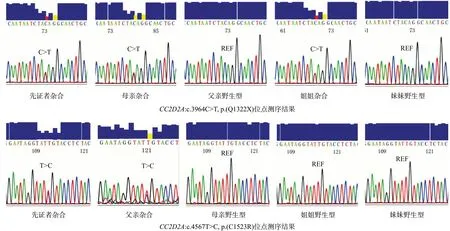
圖3 家系成員CC2D2A 基因Sanger測序結果
此外,Meckel綜合征與一些綜合征的臨床表型相互重疊,如13-三體綜合征、Bardet-Biedl綜合征等,臨床上需要進行鑒別。13-三體綜合征會出現心血管畸形及眼部異常,而很少出現肝纖維化;Bardet-Biedl綜合征中不會出現腦膨出的癥狀[5];Joubert綜合征中的典型癥狀是視網膜發育不良及舌組織腫瘤[6]。這些綜合征除了臨床表現與Meckel綜合征相互重疊以外,在患者基因組中也發現了類似Meckel綜合征的基因突變,因此從遺傳學角度講可能屬于同一類疾病。Meckel綜合征具有高度的遺傳異質性,目前已知可導致Meckel綜合征的致病基因有7個(MKS1、TMEM216、TMEM67、CEP290、RPGRIP1L、CC2D2A、NPHP3),分別位于不同的染色體上(17q22,11q13,8q22.1,12q21.32,16q12.2,4p15.32,3q22.1),并對應1~7型Meckel綜合征。因此,基因診斷也是確診Meckel綜合征的重要手段。通過影像學方法發現胎兒異常,高度懷疑為Meckel綜合征的,可收集胎兒及父母雙方的樣本提取DNA,利用高通量測序技術分析與Meckel綜合征相關的重要基因,對可疑的遺傳突變采用Sanger測序技術驗證,可明確遺傳病因并確定家系遺傳關系。2008年首次發現Meckel綜合征患者中存在CC2D2A基因的突變,在已報道的Meckel綜合征患者中,CC2D2A基因突變的占比約為13%。對于Meckel綜合征的致病機制,研究認為與細胞纖毛功能異常有關。纖毛是一種多見于脊椎動物細胞的突起結構,分布于除白細胞外的所有細胞表面,具有高度保守性,在細胞遷移和胞內外信號轉導途徑上發揮重要作用。TALLILA等[7]通過免疫熒光染色發現,CC2D2A基因突變患者的成纖維細胞中缺乏纖毛,證實了CC2D2A基因在纖毛形成中的關鍵作用。該基因發生突變可導致諸如微管形成、囊泡運輸以及信號轉導等一系列過程發生障礙的風險增加。同時,我們已知,CC2D2A基因位于人類4號染色體上,長131 692 bp,包含38個外顯子,編碼的蛋白質中含有一個卷曲螺旋狀的鈣結合結構域,此結構域在物種間高度保守,參與鈣信號通路。研究發現CC2D2A蛋白在中樞神經系統中會高度表達,其功能的缺失可導致神經發育異常,進而導致疾病的發生[8]。本研究發現的c.3964C>T(p.Q1322X)導致編碼谷氨酰胺的密碼子突變為終止密碼子,使蛋白翻譯提前終止,氨基酸序列不完整,從而使蛋白功能喪失,進而影響了纖毛功能及鈣信號通路,因此可能導致了Meckel綜合征的發病。
根據Meckel綜合征的分型和全外顯子高通量測序結果,我們認為,該先證者是由CC2D2A基因突變引起的6型Meckel綜合征。本研究發現,該先證者胎兒攜帶CC2D2A基因c.3964C>T(p.Q1322X)和c.4567T>C(p.C1523R)意義不明的復合雜合變異。在先證者母親和父親中分別發現并驗證了CC2D2A基因c.3964C>T(p.Q1322X)、c.4567T>C(p.C1523R)的雜合突變。同時,在先證者姐姐中也發現了該基因的雜合突變,此結果提示先證者中所含有的突變來源于父母,并非新發突變。CC2D2A基因c.3964C>T(p.Q1322X)突變是一種提前終止突變,該突變在EXAC數據庫和1000 human genome數據庫中的出現頻率均為0.0 001;c.4567T>C(p.C1523R)為意義不明突變位點,該突變在EXAC數據庫和1000 human genome數據庫中未有收錄。SIFT、PolyPhen-2、Mutation Taster軟件預測該位點突變均為有害。因此,我們認為CC2D2A基因c.3964C>T(p.Q1322X)和c.4567T>C(p.C1523R)意義不明復合雜合變異是該家系先證者患病的遺傳因素。此外,我們在文獻和數據庫檢索中未見該變異的相關報道,因此,本研究在臨床上首次發現了CC2D2A 基因c.3964C>T(p.Q1322X)和c.4567T>C(p.C1523R)意義不明復合雜合突變與Meckel綜合征的聯系。通過對Meckel綜合征一個相關家系的臨床及遺傳學分析明確了Meckel綜合征的臨床和基因診斷信息,CC2D2A基因變異很可能是該Meckel綜合征患病家系的致病原因,但仍需通過蛋白質組學研究進一步證實。此外,該研究重點對于已發現的遺傳病致病基因進行篩查,并不能完全排除其他少見或目前未發現的致病基因導致Meckel綜合征的可能性。我們的發現對于Meckel綜合征的確診及家系遺傳咨詢,進一步探索其發病機制、表型-基因型相關性,發現治療靶點及指導臨床工作具有重要意義。
生育過Meckel綜合征患兒的夫婦再次生育時的再發風險為25%。因此,及時發現并明確先證者患兒致病原因可為夫婦再次生育提供指導,減少出生缺陷兒的出生。胚胎植入前遺傳學診斷(preimplantation genetic diagnosis,PGD)是遺傳性出生缺陷的源頭阻斷技術。本研究的家系中先證者的致病變異及來源明確,據此可采用PGD技術阻斷該致病變異在家系中的世代遺傳。
臨床上,患有遺傳綜合征的胎兒通常表現出超聲結構異常,由于存在遺傳異質性,相同的臨床表型可能由不同的致病基因突變所致。綜合其他研究及本研究的結果,基于二代測序的WES檢測已成為對此類胎兒進行遺傳學檢測的重要策略。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
財經(2017年15期)2017-07-03 22:40:49
財經(2017年2期)2017-03-10 14:35:35
財經(2016年15期)2016-06-03 07:38:02
海峽科技與產業(2016年3期)2016-05-17 04:32:12
財經(2016年3期)2016-03-07 07:44:46
