伴有腦部損害的強直性肌營養不良10例臨床分析
2019-10-17 07:40:36胡文濤孫桂芳劉艷茹楊賀成
中風與神經疾病雜志 2019年9期
關鍵詞:信號
胡文濤,孫桂芳,劉 希,劉艷茹,王 賞,彭 濤,楊賀成,盧 宏
強直性肌營養不良癥(DM)是常染色體顯性遺傳的進行性多系統損害的疾病,分為DM1型和DM2型,其中DM1型在成人中最常見,是由19號染色體上的DMPK基因3’端非編碼區CTG三核苷酸重復序列的動態突變引起的[1],DM2則是由鋅指蛋白9(ZNF9)基因的CCTG序列擴增引起。除了常見的骨骼肌強直、無力和萎縮外,DM常累及多系統損害,其中累及中樞神經系統者可出現智力低下、嗜睡等癥狀,頭部MRI可見顱骨內板增厚、蝶鞍變小、腦萎縮、局部或彌漫性腦白質損害[2]。國內DM患者很少行常規頭部影像學檢查,關于DM腦部的異常鮮有報道。本研究總結分析10例伴有腦部損害的DM患者的臨床資料,探討其臨床特點,提高對此病的認識水平。
1 資料與方法
1.1 一般資料 為2012年4月~2018年4月鄭州大學第一附屬醫院就診的DM患者,共檢出行頭部MRI有腦部損害的10例患者,其中男性7例,女性3例,發病年齡13~48歲,家族中有類似病史表現者6例,其中1例患者姐姐已確診。
1.2 研究方法 回顧性分析10例患者的臨床資料,包括臨床表現、頭部MRI表現、肺部檢查、神經肌電圖、肌肉MRI、肌肉活檢及基因學檢查。
2 結 果
2.1 主要臨床表現 見表1。本組患者均為慢性起病,以四肢無力、僵硬、不靈活起病7例,以言語緩慢、不利起病者2例,以肌強直為主者1例。肌無力以四肢遠端為主,肌強直以雙手虎口區為主,叩診可見肌球。肌肉萎縮者2例,1例位于左肩胛區,另1例為雙上肢。出現智能損害者7例,白天嗜睡者1例,疲乏感者1例,情緒低落者2例,禿頂者2例,心臟受累者3例,確診白內障者2例,性功能障礙者1例,呼吸道感染、呼吸困難行氣管插管者1例,構音障礙者6例。
2.2 神經電生理檢查 見表1。10例患者均行肌電圖檢測,均提示肌源性損害,且均有肌強直電位,主要出現在脛前肌、腓腸肌、拇對掌肌、三角肌及股內側肌等肌肉。神經電圖:2例出現周圍神經傳導速度減慢,3例出現雙脛神經H反射未引出,1例出現感覺神經電位波幅降低,1例出現運動神經電位波幅降低。運動誘發電位:2例出現雙下肢錐體束未引出。感覺誘發電位:1例出現雙下肢深感覺傳導通路潛伏期延遲。
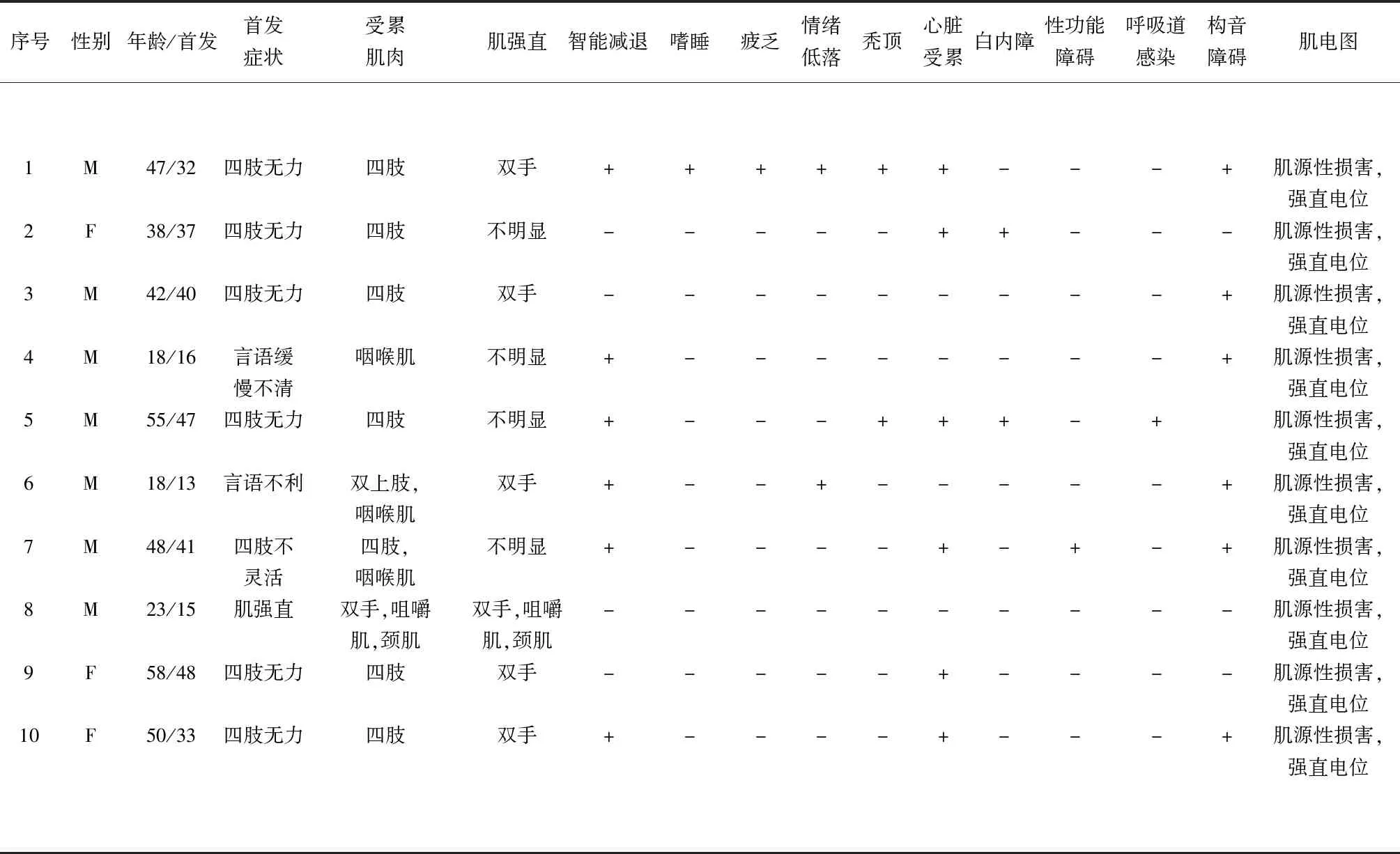
表1 10例DM患者基本臨床表現
2.3 影像學檢查 見表2。10例患者均行頭部MRI檢查,全部出現腦部異常,其中9例為白質異常信號,1例為幕上腦室積水,<18歲者2例,2例>50歲。雙側額頂顳葉白質高信號者9例,雙側腦室旁白質高信號1例,左側島葉白質高信號1例,均考慮為脫髓鞘病變;3例見顱板增厚,2例出現側腦室增大或幕上腦積水(見圖1),1例見輕度腦萎縮,1例見前床突腦膜瘤,1例示腦部灰質異常信號,2例同時行脊髓MRI分別發現頸胸脊髓異常信號。3例行肌肉MRI,均顯示肌肉組織出現片狀壓脂高信號,提示受累肌群組織水腫,其中1例出現雙側小腿肌束、肌腹變細,提示肌肉萎縮。1例患者行肺部CT示雙肺多發條絮樣高密度影,右肺下葉實變,提示肺部感染,雙側胸腔積液。
2.4 病理學檢查 3例患者行骨骼肌活檢,活檢部位為右肱二頭肌、右側三角肌及左股四頭肌。3例均出現肌束衣內結締組織或脂肪組織增生(見圖2),1例可見少量淋巴細胞浸潤;肌束內均可見較多的萎縮及肥大肌纖維,可見較多核內移;2例可見核聚集,1例可見肌漿塊;1例NADH和SDH染色可見較多蟲蝕肌纖維及靶樣纖維;1例NADH和SDH染色見部分肌纖維氧化酶分布不均勻;1例ORO染色見肌滴增多,1例PAS染色見部分肌纖維內糖原缺失,ATP酶、MHCs和MHCf染色均可見萎縮肌纖維累及兩型,以I型為主。
2.5 基因學檢查 2例患者采用聚合酶鏈反應(PCR)方法,針對DMPK的三核苷酸重復序列進行PCR 擴增及毛細管電泳,分析其片段大小,計算三核苷酸重復次數,從而確定是否患病。1例患者樣本DMPK基因(CTG)拷貝數n=116,遠超過正常范圍(正常n<37),為DM1型(見圖3),另1例患者DMPK基因(CTG)拷貝數n>50,提示DM1型。

表2 10例DM患者影像學表現
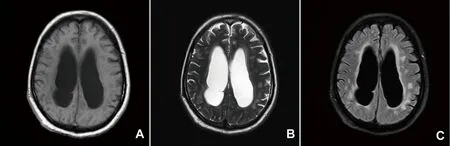
圖1 頭部MRI提示:雙側側腦室旁白質脫髓鞘,腦積水(男,表現為四肢無力,智能下降,無錐體束征,無血管疾病的證據)A:T1相;B:T2相;C:T2flair相
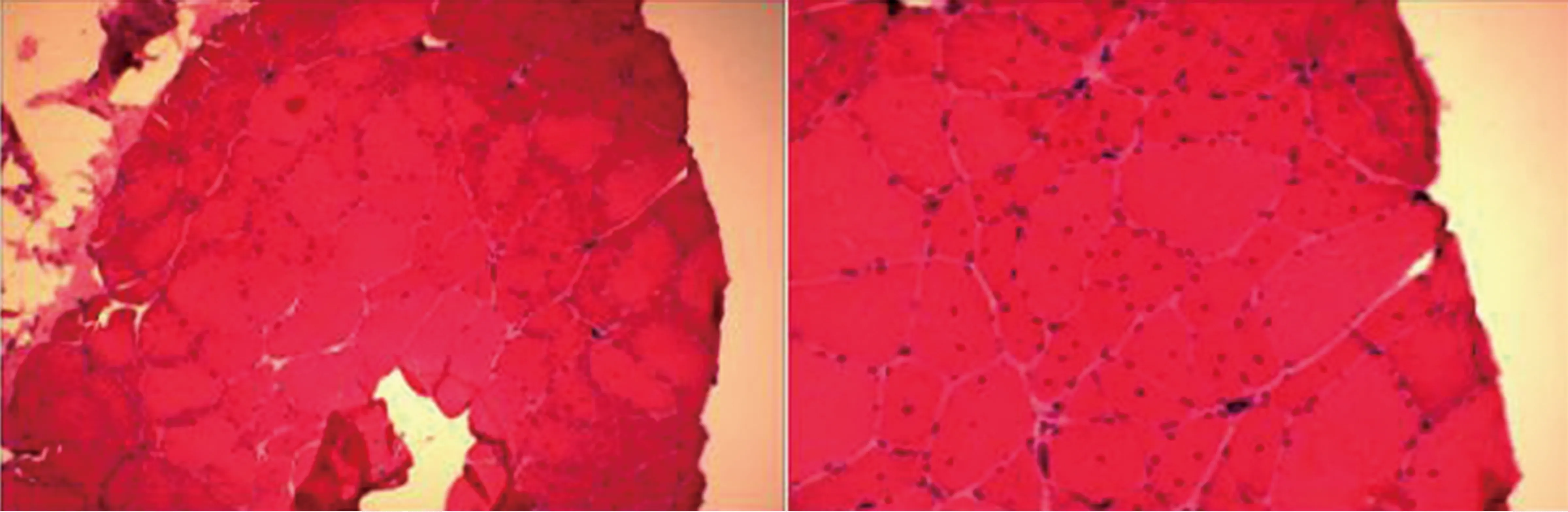
圖2 肌肉活檢提示:右側三角肌HE及GT染色肌束衣內結締組織及脂肪組織稍增生,肌束內局部肌內衣增生,肌纖維大小不均勻,可見萎縮及肥大肌纖維,可見核內移增加和固縮核團塊,NADH和SDH染色示部分肌纖維氧化酶分布不均勻,ATP酶染色顯示萎縮肌纖維累及兩型,以I型為主。
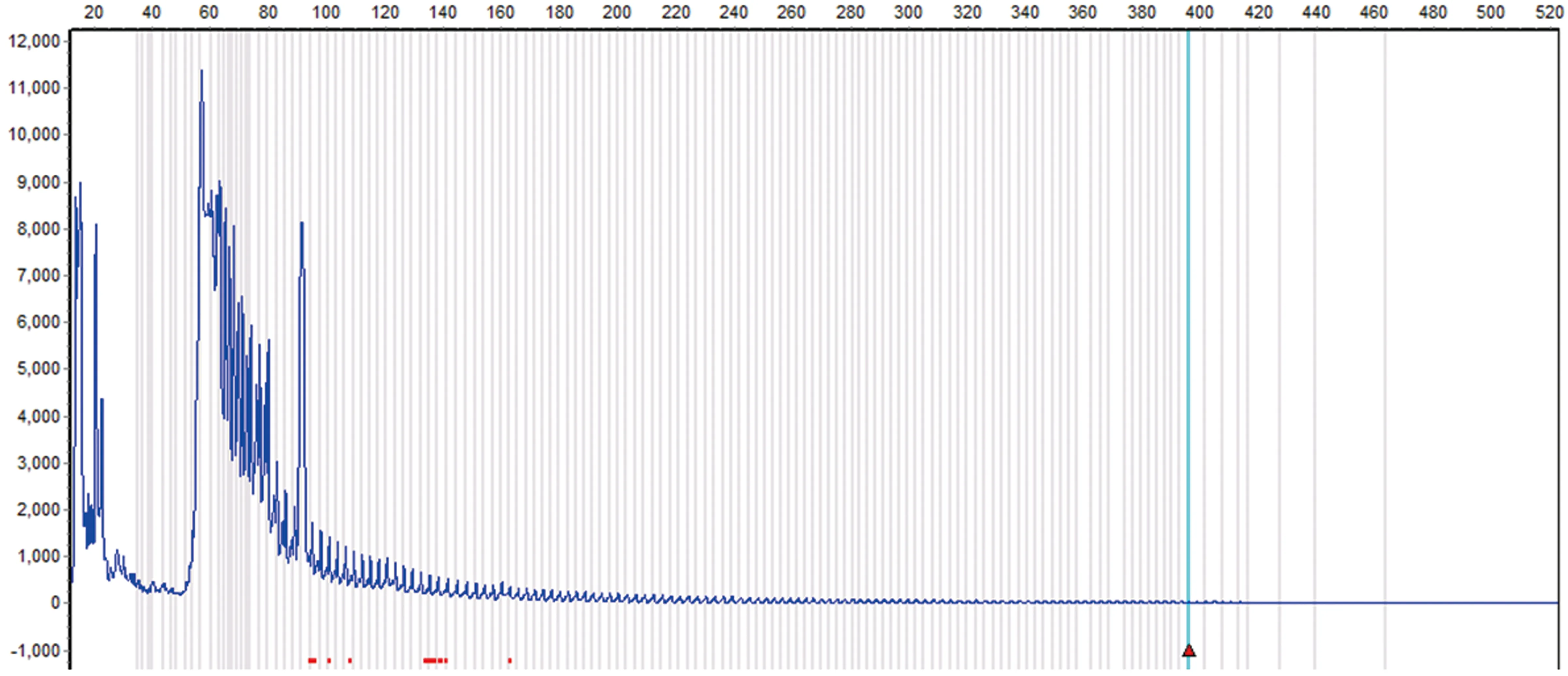
圖3 患者樣本DMPK基因(CTG)拷貝數n=116,超過正常范圍(正常n<37),為DM1型
3 討 論
DM是一種累及多系統的常染色體顯性遺傳病,是成人肌營養不良中最常見的類型[3]。根據臨床癥狀和基因分型,DM分為DM1型和DM2型兩種。DM發病的分子機制非常復雜,目前關于基因異常如何導致DM的發生尚存在爭議,以DM1為例,DMPK蛋白表達減少、DMPK基因表達異常以及異常轉錄生成的毒性mRNA均可導致DM1的發生[4]。本組患者因條件限制,只有2例行基因檢測,確診為DM1型,其余尚無法確定其DM分型。在這兩例DM1患者中,1例患者家族中并無相似患者,推測其為基因突變所致,相對與本組其他患者,除肌無力、肌強直外,其累及的其他系統癥狀較廣泛,包括嗜睡、智能減退、日間疲乏、情緒低落、禿頂、心臟異常和構音障礙等。
DM患者的主要臨床表現為肌無力、肌強直、肌萎縮,肌強直是本病的主要特征。本組10例患者中以四肢無力、僵硬、不靈活起病7例,以肌強直為主者1例。肌無力以四肢遠端為主,肌強直以雙手虎口區為主,叩診可見肌球。肌肉萎縮者2例,分別位于左肩胛區和雙上肢。肌電圖是診斷DM的重要檢查手段,本組患者肌電圖檢測均提示肌源性損害,且均伴有肌強直電位,主要出現在脛前肌、腓腸肌、拇對掌肌、三角肌及股內側肌等肌肉。除了肌肉受累以外,神經電圖及運動和感覺誘發電位也出現異常,提示DM不但侵犯肌纖維,有可能同時侵犯了神經纖維和傳導束。
DM本質上是多系統疾病,中樞神經系統也會同時受累[5]。成人DM1患者往往有執行能力和視空間能力的障礙,DM2患者除了上述障礙,還有口頭記憶能力的障礙[6]。隨著影像學的進步,中樞神經系統損害被越來越多的發現,頭部MRI的異常表現多樣,包括白質高信號、腦室擴大、腦萎縮等,其中白質高信號最為常見,發生率約為78%[7]。以往研究發現DM1患者的頭部MRI檢查中,除了白質高信號以外,還伴隨著灰質和白質體積的減少,而在DM2患者中除了相同的白質損害外,還存在更加顯著的灰質損害[5,8~10]。白質纖維束的損害和復雜神經網絡結構的紊亂與DM患者的認知和行為異常存在著一定程度的聯系[10~12],在伴有白質損害DM1患者中,其白質高信號與患者的記憶、執行、視空間和推理能力的損害密切相關[13]。DM患者腦部損害發病機制目前尚不完全清楚,研究表明神經細胞內異常的微管相關tau蛋白的聚集造成的神經纖維變性與DM1的腦部損害密切相關[14~16]。本組10例患者均行頭部MRI檢查,全部出現腦部異常,以白質異常信號為主,同時還存在腦室擴大、積水、腦膜瘤、板障增厚、灰質異常信號及腦萎縮等異常。本組多例患者癥狀上存在智能減退、嗜睡,白天和情緒低落可能與其腦部損害有關。
DM患者呼吸肌的受累會導致呼吸道的清除功能受損,肺泡低通氣合并膈肌無力使患者排痰困難,再加上肌肉組織營養不良,常發生食管誤吸,導致患者肺部感染,疾病后期常常出現呼吸衰竭[17]。本組1例患者后期發生呼吸道感染,繼而發生呼吸道衰竭,給予氣管插管,呼吸機輔助呼吸及抗感染治療,后病情好轉轉至當地醫院治療,早期的肺功能檢測有助于早期發現呼吸肌無力,對患者的后期治療有一定的幫助。此外,本組3例患者行肌肉MRI,均顯示肌肉組織出現片狀壓脂高信號,提示受累肌群組織水腫,可能與基因突變導致炎性細胞浸潤有關[18],肌肉MRI的應用有助于DM的診斷。
綜上所述,DM是一種多系統累及的遺傳性疾病,除肌無力、肌強直和肌萎縮外,還常累及腦部白質、腦室腦膜、呼吸等其他系統,對診斷DM的患者進行頭部和呼吸系統檢查,有助于早期發現患者的腦損害和肺功能異常并進行早期干預;此外,肌肉MRI對DM的診斷有一定幫助。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
