紫外吸收波長對人纖維蛋白含量測定的影響
2019-08-01 01:26:14于濤孫麗麗
吉林農(nóng)業(yè) 2019年14期
關(guān)鍵詞:測定
于濤 孫麗麗
摘要:本文探索了高效液相色譜法檢測人纖維蛋白含量的色譜條件,通過對人纖維蛋白溶液紫外光譜的掃描結(jié)果,設定不同波長進行比對,并篩選出適合的波長,為開展進一步分析提供理論依據(jù)。
關(guān)鍵詞:紫外光譜;人纖維蛋白含量;測定
中圖分類號: R927 ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ?文獻標識碼: ?A ? ? ? ? ? ? ? ? ? ? ? ? DOI編號: 10.14025/j.cnki.jlny.2019.14.031
高效液相色譜法和氨基酸分析儀是目前蛋白質(zhì)分析方法中最有效的手段,高效液相色譜法分析原理主要是利用蛋白質(zhì)的氨基與標識化試劑作柱前或柱后衍生,再進行測定。目前,用于反相高效液相色譜檢測氨基酸的柱前衍生試劑近10種,以柱前衍生高效液相色譜法測定纖維蛋白原中的精氨酸也有報道,但其衍生化試劑盒費用高,且需配置熒光檢測器。本實驗參照 《中國藥典》二部(2005版)中高效液相色譜法,采用2,4-二硝基氟苯為柱前衍生劑,以紫外吸收檢測器檢測不同波長的吸收效果,選出最佳的吸收波長。
1 材料與方法
1.1 樣品準備
人纖維蛋白原為BIOSHARP公司產(chǎn)品,規(guī)格:100mg/支,Exp:2017/03。
1.2 主要試劑及儀器
磷酸二氫鈉,磷酸氫二鈉,乙腈(色譜純),凝膠色譜柱Shodex KW-802.5,島津紫外檢測器,普析TU-1901型紫外分光光度計。
1.3 流動相及樣品溶液的制備
流動相采用乙腈-磷酸鹽緩沖溶液,稱取磷酸二氫鈉9.5g,磷酸氫二鈉1.25g,置于500mL容量瓶中,超純水溶解并定容,另取500mL乙腈與其混合,用0.45μm有機膜過濾。
1.4 樣品處理
精密量取樣品溶液1mL,加入相對應的流動相乙腈-磷酸鹽緩沖液和乙腈-純水溶液99mL,充分混勻,靜止30min,進行蛋白質(zhì)沉淀,離心5min,取上層清液進行測定。
1.5 色譜條件
采用凝膠色譜柱Shodex KW-802.5(5μm,8*300 mm);流速1mL/min;波長設定為205nm、215nm、230nm、280nm;柱溫40℃;采用島津LC-15C色譜工作站;進樣量20μL。
1.6 實驗方法
分別在不同波長下分析蛋白分子保留時間及色譜峰吸收強度,篩選最適合的實驗條件。
2 實驗結(jié)果
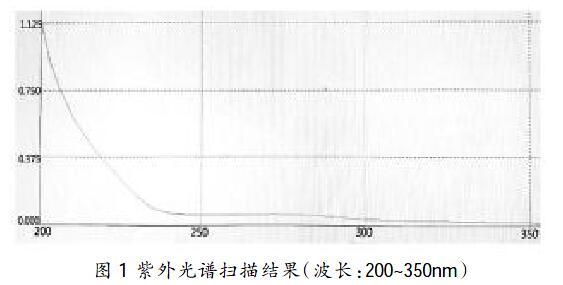
掃描紫外光譜,波長200~350nm,結(jié)果見圖1。
通過掃描紫外光譜可得出蛋白溶液200~230nm內(nèi)有較強吸收強度,在250~300nm有較弱的吸收強度,故設定檢測波長為205nm、215nm、230nm、280nm。
不同吸收波長下檢測結(jié)果(流動相配比為乙腈:磷酸鹽緩沖液=1:1)。
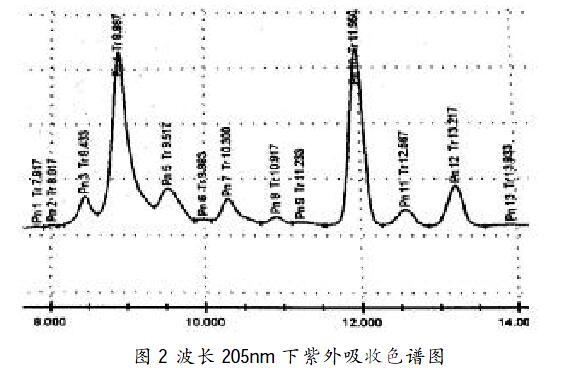
在波長為205nm下進行檢測,結(jié)果見圖2。
由色譜圖結(jié)果可得出,色譜峰的吸收強度不高,各氨基酸組分分離度≤0.5,色譜峰存在一定拖尾現(xiàn)象,分離效果不理想。
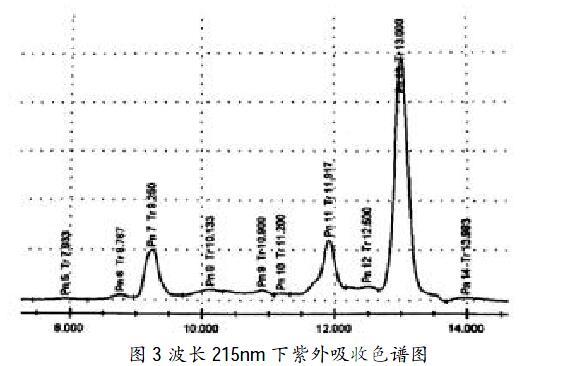
在波長215nm下進行檢測,結(jié)果見圖3。
由色譜圖結(jié)果可得出,色譜峰的吸收強度較好,各氨基酸組分分離度≥1.5,色譜峰對稱性理想,分離效果較為滿意。
在波長230nm下進行檢測,結(jié)果見圖4。
由色譜圖結(jié)果可得出,色譜峰的吸收強度降低,各氨基酸組分分離度≤1,色譜峰對稱性不理想,且蛋白組分比例發(fā)生明顯變化,分離效果不滿意,不建議在此波長下分析檢測。
在波長280nm下進行檢測,結(jié)果見圖5。
由色譜圖結(jié)果可得出,色譜峰的吸收強度大幅降低,各氨基酸組分分離度≤0.5,色譜峰對稱性很不理想,且蛋白組分無法分離,此波長不適合進行蛋白質(zhì)的分析檢測。
3 結(jié)論
本實驗建立的色譜方法采用凝膠色譜柱進行分離,通過多次單因素實驗,確定在215nm波長下蛋白分子有最佳吸收強度和分離度,通過對此次實驗方法條件的摸索,得到最適合吸收波長實驗條件,測定結(jié)果穩(wěn)定,有良好的重復性,可直接用于人纖維蛋白含量測定,用于進一步實驗研究。
參考文獻
[1]尉小平,冉鐵成,王堅,趙家宏.柱前衍生反相高效液相色譜法測定凍干人纖維蛋白原制品中鹽酸精氨酸和甘氨酸的含量[J].中國生物制品學,2007,21:645-687.
[2]陳通威,高紅琴,崔素梅,張惠蘭,王宗剛.高效液相色譜法測定人纖維蛋白原中甘氨酸、鹽酸精氨酸含量[J].中國藥業(yè),2013,31:635-637.
[3]候淑琴,梁小明,黃玲,張惠蘭,張麗玲.人纖維蛋白原制劑中蛋白質(zhì)含量測定方法的探究[J].臨床醫(yī)藥實踐,2014,21:521-545.
作者簡介:于濤,碩士,中級講師,研究方向:玉米深加工、儀器分析。
猜你喜歡
農(nóng)機使用與維修(2016年12期)2017-01-17 17:30:33
安徽農(nóng)學通報(2016年24期)2017-01-12 02:10:04
科學與財富(2016年29期)2016-12-27 16:45:09
科學與財富(2016年29期)2016-12-27 16:43:09
科學與財富(2016年29期)2016-12-27 16:42:10
綠色科技(2016年20期)2016-12-27 16:04:51
東方教育(2016年3期)2016-12-14 20:35:31
中國科技博覽(2016年13期)2016-07-13 01:51:35
中國科技博覽(2016年11期)2016-05-06 04:30:49
中國科技博覽(2016年10期)2016-04-29 03:36:52
