鹽酸小檗堿-磷脂復(fù)合物的處方工藝研究
2019-07-18 11:07:16王慶劉黎瑤通信作者王慶奎
天津農(nóng)學(xué)院學(xué)報 2019年2期
王慶,劉黎瑤, ,通信作者,王慶奎
鹽酸小檗堿-磷脂復(fù)合物的處方工藝研究
王慶a,劉黎瑤a, b,通信作者,王慶奎b
(天津農(nóng)學(xué)院,a. 基礎(chǔ)科學(xué)學(xué)院 生物制藥系;b. 天津市水產(chǎn)生態(tài)及養(yǎng)殖重點實驗室,天津 300384)
鹽酸小檗堿是臨床治療胃腸道細(xì)菌感染等疾病的有效藥物。但由于脂溶性差等問題,造成其口服生物利用度較低,難以充分發(fā)揮藥效。本研究旨在利用磷脂復(fù)合技術(shù)提高鹽酸小檗堿的脂溶性。通過溶劑-旋轉(zhuǎn)蒸發(fā)法,以復(fù)合率()為篩選指標(biāo),采用單因素篩選及正交設(shè)計,篩選得到制備鹽酸小檗堿-磷脂復(fù)合物的最佳處方。優(yōu)選處方下,鹽酸小檗堿-磷脂復(fù)合率達到90%以上,4 ℃下可穩(wěn)定保存3個月。該研究為鹽酸小檗堿-磷脂復(fù)合物的后期應(yīng)用奠定了物質(zhì)基礎(chǔ)。
鹽酸小檗堿;磷脂復(fù)合物;正交設(shè)計;處方篩選
鹽酸小檗堿作為中藥黃柏、黃連中的有效成分,是臨床治療胃腸道細(xì)菌感染等疾病的有效藥物。并且,有大量文獻報道其在治療腫瘤及心腦血管疾病方面有效性較高,具有較高的潛在開發(fā)價值[1-3]。但由于鹽酸小檗堿的脂溶性極差,導(dǎo)致口服給藥后的生物利用度較低,無法充分發(fā)揮藥物治療作用。在口服給予鹽酸小檗堿時,如果要達到治療心腦血管疾病的血藥濃度,勢必要增加給藥劑量,繼而產(chǎn)生諸多不良反應(yīng),如腸道菌群失衡、產(chǎn)生耐藥性等[4-5]。因此,通過藥物制劑技術(shù)構(gòu)建適宜的遞送載體來提高鹽酸小檗堿的脂溶性,具有極大的應(yīng)用前景和價值。
磷脂復(fù)合物通過藥物和磷脂分子間的靜電復(fù)合作用形成穩(wěn)定的復(fù)合體,能夠在不改變藥物分子結(jié)構(gòu)的前提下,極大程度地提高藥物的脂溶性,在增加中藥有效成分的口服生物利用度方面已有較深入的研究[6-8]。已有研究將鹽酸小檗堿制備成磷脂復(fù)合物,但其復(fù)合時間長達2~4 h,而復(fù)合率基本在70%左右[9]。本研究中發(fā)現(xiàn),輔助利用超聲的方法能極大縮短復(fù)合時間,且通過復(fù)合溶劑篩選,選擇適宜的復(fù)合溶劑條件,可大幅度增加復(fù)合率。經(jīng)單因素篩選及正交設(shè)計,以鹽酸小檗堿與大豆卵磷脂的復(fù)合率為篩選指標(biāo),對復(fù)合溶劑、超聲復(fù)合時間以及投料比例進行了考察,優(yōu)選復(fù)合處方工藝中,復(fù)合溶劑為甲醇-二氯甲烷系統(tǒng),溶劑系統(tǒng)的最適pH值為6.0,超聲復(fù)合時間為30 min,鹽酸小檗堿與磷脂的投料質(zhì)量比例為1∶1。按照優(yōu)選處方所制備的鹽酸小檗堿-磷脂復(fù)合物的復(fù)合率達到90%以上。
1 儀器與試劑
1.1 儀器
電子天平(Sartorius,BSA 223S);超聲波清洗儀(昆山禾創(chuàng)儀器有限公司,KH5200B型);紫外可見分光光度計(北京普析通用儀器有限公司);旋轉(zhuǎn)蒸發(fā)儀(Rotavapor R-100,BUCHI);微孔濾膜等。
1.2 試劑
鹽酸小檗堿(西安奧賽生物科技有限公司,純度99%);鹽酸小檗堿標(biāo)準(zhǔn)品(中國藥品生物制品檢定院,HPLC≥98%);大豆磷脂(上海艾偉拓醫(yī)藥科技有限公司,純度98%);無水乙醇、正己烷、甲醇、四氫呋喃、二氯甲烷(天津市風(fēng)船化學(xué)試劑有限公司,分析純);磷酸、氫氧化鈉等其他試劑均為分析純。
2 試驗方法
2.1 鹽酸小檗堿-磷脂復(fù)合物的制備方法
分別稱取適量鹽酸小檗堿與大豆卵磷脂置于燒杯中,加入適宜溶劑分別溶解后,混合并振搖均勻。置于超聲條件下進行復(fù)合反應(yīng)。超聲復(fù)合結(jié)束后,減壓蒸發(fā)多余溶劑,即得干燥的黃色固體鹽酸小檗堿-磷脂復(fù)合物。
2.2 鹽酸小檗堿-磷脂復(fù)合物的復(fù)合率測定方法建立
2.2.1 紫外-可見光區(qū)全波長掃描
稱取鹽酸小檗堿標(biāo)準(zhǔn)品10 mg置于容量瓶中,加入無水乙醇超聲溶解并稀釋至適當(dāng)濃度后,使用紫外-可見分光光度計測定其在200~900 nm波長區(qū)域內(nèi)的紫外-可見光吸收情況,并記錄該區(qū)域內(nèi)的掃描光譜圖。根據(jù)全波長掃描光譜圖選擇含量測定的最適波長。
2.2.2 標(biāo)準(zhǔn)曲線繪制
準(zhǔn)確稱量鹽酸小檗堿標(biāo)準(zhǔn)品10 mg置于容量瓶中,加入無水乙醇超聲溶解并定容至10 mL,配制得到濃度為1 000 μg/mL的儲備液。精密移取適量儲備液,并使用無水乙醇稀釋得到20、40、60、80、100 μg/mL梯度濃度的標(biāo)準(zhǔn)溶液。通過紫外分光光度法,在最適吸收波長條件下,測定上述系列標(biāo)準(zhǔn)溶液的吸光度。以標(biāo)準(zhǔn)溶液的質(zhì)量濃度為橫坐標(biāo),以吸光度為縱坐標(biāo),繪制標(biāo)準(zhǔn)曲線,并通過最小二乘法擬合出線性回歸方程。
2.2.3 精密度測定
取2.2.2部分低、中、高濃度的線性樣品連續(xù)測定5次,測定其在最適波長下的吸光度,記錄吸光度數(shù)值并計算相對標(biāo)準(zhǔn)偏差(RSD%)。
2.2.4 復(fù)合率的測定方法
利用鹽酸小檗堿不溶于正己烷,而成功形成鹽酸小檗堿-磷脂復(fù)合物后則易溶于正己烷的特性[10-11],在樣品制備結(jié)束后,稱取適量樣品置于燒杯中,加入適量正己烷溶解,經(jīng)微孔濾膜過濾后,收集濾液并減壓蒸發(fā)去除多余正己烷,使用無水乙醇溶解并稀釋適宜倍數(shù),于最適吸收波長條件下,測定其中鹽酸小檗堿的吸光度,使用標(biāo)準(zhǔn)曲線的回歸方程,計算復(fù)合物中鹽酸小檗堿的含量。根據(jù)初始投料量(0)與復(fù)合物中鹽酸小檗堿的質(zhì)量(t)計算復(fù)合率(),計算公式為:(%)=t/0×100%。
2.3 單因素影響試驗
2.3.1 反應(yīng)溶劑篩選
根據(jù)文獻調(diào)研[8]和前期試驗結(jié)果,本研究首先對復(fù)合反應(yīng)的溶劑系統(tǒng)進行了篩選。設(shè)定了鹽酸小檗堿與磷脂的投料比為1∶10(質(zhì)量比例),超聲復(fù)合時間為30 min。以鹽酸小檗堿-磷脂復(fù)合物的復(fù)合率為評價指標(biāo),對甲醇、乙醇、二氯甲烷、四氫呋喃進行考察,以篩選出最佳的反應(yīng)溶劑。
2.3.2 溶劑系統(tǒng)pH值篩選
選定反應(yīng)溶劑后,對該反應(yīng)溶劑系統(tǒng)的pH值進行篩選,在篩選過程中,保持鹽酸小檗堿與磷脂的投料比為1∶10(質(zhì)量比例),超聲復(fù)合時間為30 min。設(shè)定溶劑系統(tǒng)的pH值為2.0、4.0、6.0、8.0、12.0,以復(fù)合率為評價指標(biāo),篩選出反應(yīng)溶劑系統(tǒng)的最佳pH值。
2.3.3 投料比例篩選
使用選定的反應(yīng)溶劑,且在選定的pH條件下,保持超聲復(fù)合反應(yīng)30 min。以鹽酸小檗堿-磷脂復(fù)合物的復(fù)合率為評價指標(biāo),設(shè)定鹽酸小檗堿與磷脂的投料比為1∶1、1∶3、1∶5、1∶10、1∶15(質(zhì)量比例),對鹽酸小檗堿與磷脂的投料比例(質(zhì)量比例)進行篩選。
2.3.4 超聲反應(yīng)時間篩選
使用選定的反應(yīng)溶劑,且在選定的pH條件及投料比例下,以鹽酸小檗堿-磷脂復(fù)合物的復(fù)合率為評價指標(biāo),設(shè)定超聲時間分別為15、30、60、90、120 min,對復(fù)合過程的超聲時間進行篩選。
2.4 正交設(shè)計試驗
根據(jù)單因素篩選結(jié)果,選擇最佳反應(yīng)溶劑系統(tǒng)為甲醇-二氯甲烷,復(fù)合反應(yīng)受到溶劑系統(tǒng)pH值(A)、鹽酸小檗堿與磷脂的投料比例(B)、超聲時間(C)的影響。因此,進行上述3因素3水平的正交試驗,以復(fù)合率為指標(biāo)篩選出最佳的處方工藝。正交設(shè)計因素水平表如表1所示。
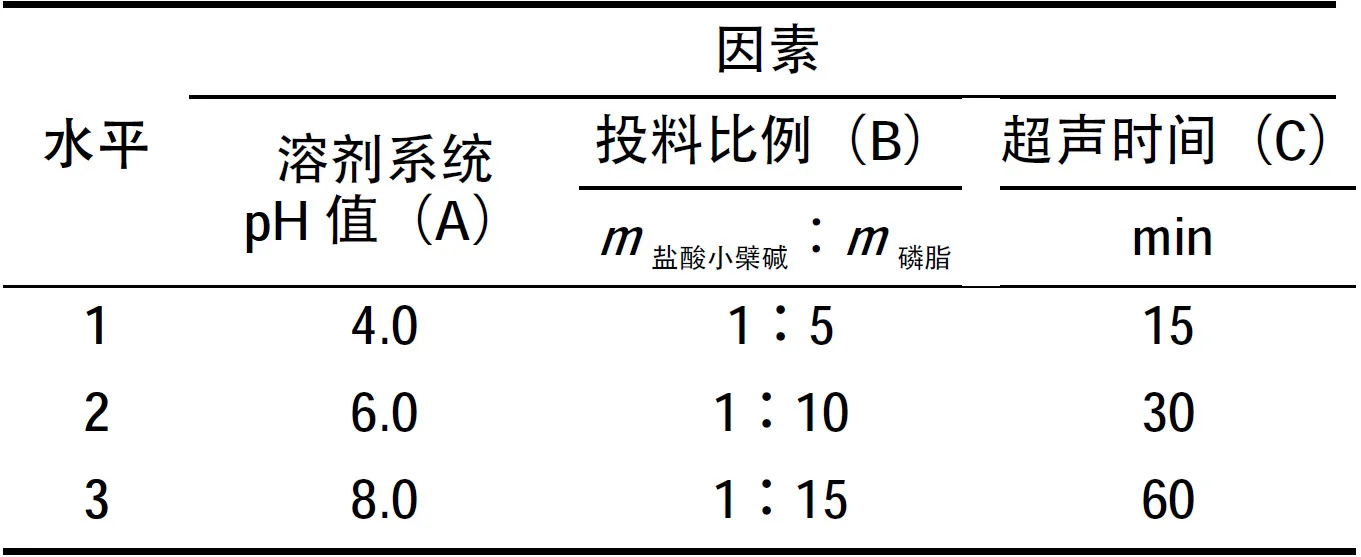
表1 正交設(shè)計因素水平表
2.5 優(yōu)化處方下復(fù)合物的放置穩(wěn)定性
在優(yōu)選的處方工藝下,制備3批鹽酸小檗堿-磷脂復(fù)合物,分別測定其復(fù)合率,且于4 ℃條件下密封放置0、1、2、3個月,對該優(yōu)選處方下放置穩(wěn)定性進行評價。
3 結(jié)果與分析
3.1 鹽酸小檗堿的含量測定方法
3.1.1 全波長掃描
按照既定試驗方法,使用紫外-可見分光光度計測定鹽酸小檗堿溶液在200~900 nm波長區(qū)域內(nèi)的紫外-可見光吸收情況,并記錄了該區(qū)域內(nèi)的掃描光譜圖,如圖1所示。根據(jù)全波長掃描光譜圖選擇含量測定的最適波長為350 nm。
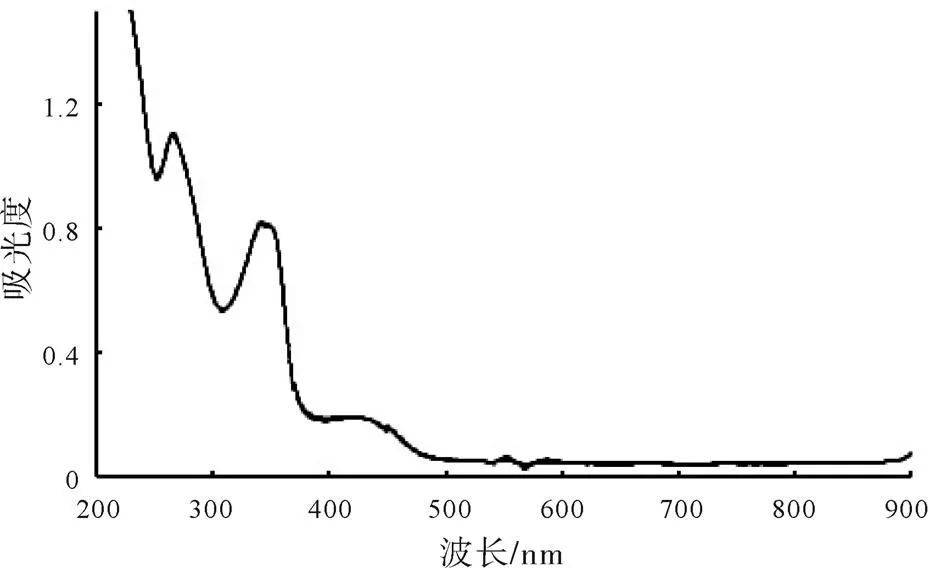
圖1 鹽酸小檗堿溶液的全波長掃描光譜圖
3.1.2 標(biāo)準(zhǔn)曲線繪制
按照既定的試驗方法,配制了不同濃度鹽酸小檗堿標(biāo)準(zhǔn)品系列溶液,依法使用紫外分光光度法進行測定。結(jié)果表明:以吸光度Abs(縱坐標(biāo),)對濃度(μg/mL,橫坐標(biāo),)進行線性回歸,擬合得出的回歸方程為=0.008 9+0.025 2(2= 0.999 8),鹽酸小檗堿標(biāo)準(zhǔn)品系列溶液在0~100 μg/mL濃度范圍內(nèi)線性關(guān)系良好,如圖2所示。
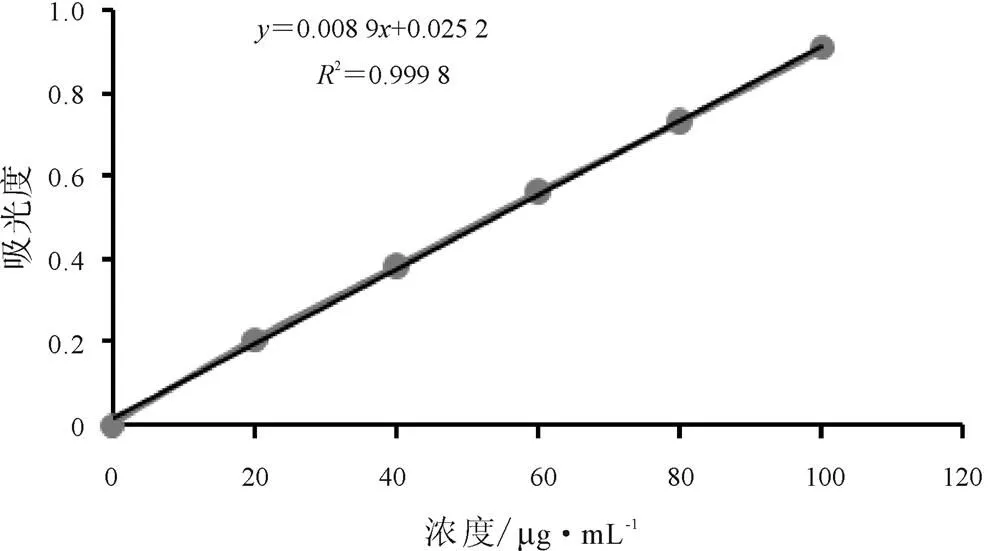
圖2 鹽酸小檗堿標(biāo)準(zhǔn)品系列溶液標(biāo)準(zhǔn)曲線
3.1.3 精密度測定
按照既定的試驗方法,低、中、高濃度鹽酸小檗堿標(biāo)準(zhǔn)品溶液的精密度測定結(jié)果如表2所示。

表2 精密度試驗結(jié)果
注:為待測溶液在350 nm處的吸光度數(shù)值;為標(biāo)準(zhǔn)偏差;(%)為相對標(biāo)準(zhǔn)偏差
測定結(jié)果表明,對低、中、高濃度對照品溶液分別連續(xù)重復(fù)5次測定的值均小于2%,表明本法的精密度良好。
3.2 單因素影響試驗
3.2.1 反應(yīng)溶劑篩選
根據(jù)既定試驗方法,以鹽酸小檗堿-磷脂復(fù)合物的復(fù)合率為評價指標(biāo),對甲醇、乙醇、二氯甲烷、四氫呋喃進行考察,以篩選出最佳的反應(yīng)溶劑,篩選結(jié)果如圖3所示。分析結(jié)果表明,使用甲醇-二氯甲烷的溶劑系統(tǒng)制備鹽酸小檗堿-磷脂復(fù)合物時,復(fù)合率均高于其他幾種溶劑,因此,在后續(xù)處方篩選中,均以甲醇-二氯甲烷作為反應(yīng)溶劑。
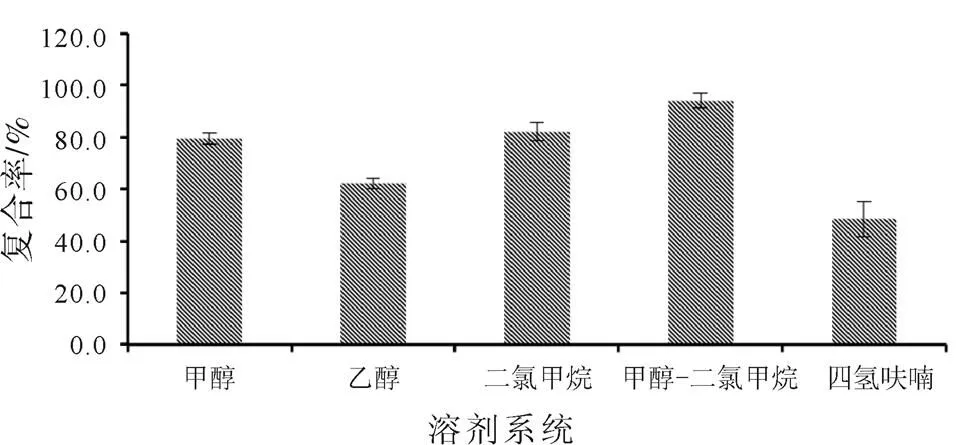
圖3 反應(yīng)溶劑篩選結(jié)果
3.2.2 溶劑系統(tǒng)pH值篩選
根據(jù)既定試驗方法,在篩選過程中保持鹽酸小檗堿與磷脂的投料比為1∶10(質(zhì)量比例),超聲復(fù)合時間為30 min,并以甲醇-二氯甲烷作為復(fù)合溶劑。以復(fù)合率為評價指標(biāo),篩選出反應(yīng)溶劑系統(tǒng)的最佳pH值,篩選結(jié)果如圖4所示。分析結(jié)果表明,當(dāng)反應(yīng)溶劑系統(tǒng)的pH值從2.0升高至6.0時,復(fù)合率逐漸升高;pH為6.0時復(fù)合率達到最高,隨后便開始降低。
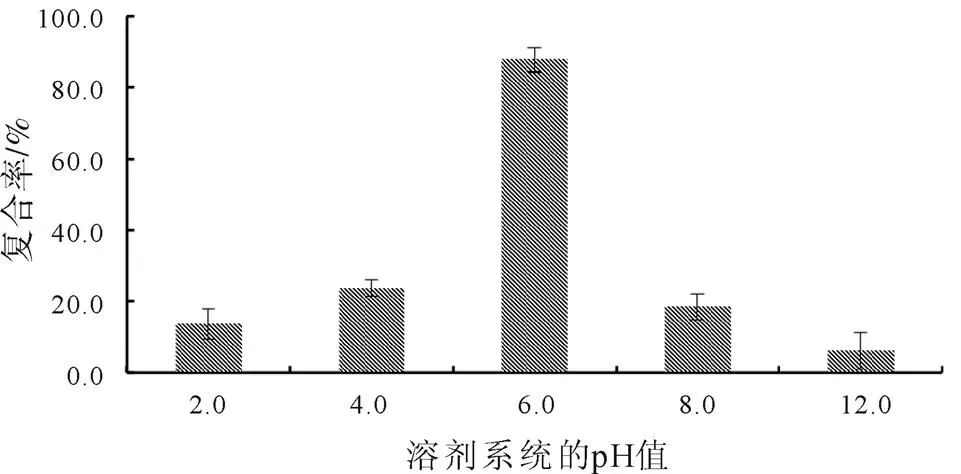
圖4 溶劑系統(tǒng)pH值的單因素篩選結(jié)果
3.2.3 投料比例篩選
根據(jù)既定試驗方法,保持溶劑系統(tǒng)的pH值為6.0、超聲復(fù)合反應(yīng)為30 min條件下,以鹽酸小檗堿-磷脂復(fù)合物的復(fù)合率為評價指標(biāo),對鹽酸小檗堿與磷脂的投料比例(質(zhì)量比例)進行篩選,結(jié)果如圖5所示。分析結(jié)果表明,隨著磷脂用量的增加,復(fù)合率呈現(xiàn)出增長的趨勢,并且在1∶10之后逐漸趨近于復(fù)合平衡,復(fù)合率基本達到90%左右。
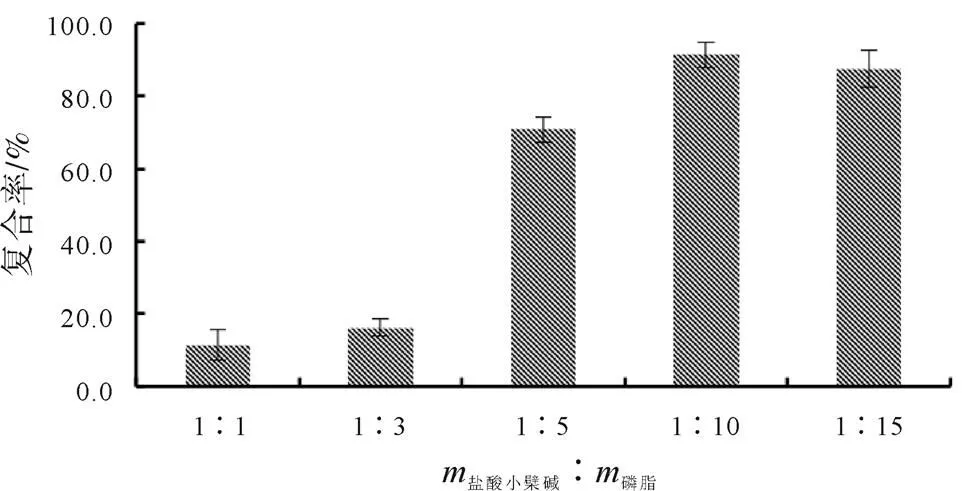
圖5 鹽酸小檗堿與磷脂的投料比例(質(zhì)量比例)單因素篩選結(jié)果
3.2.4 超聲反應(yīng)時間篩選
在選定的反應(yīng)溶劑、選定的pH條件以及投料比例下,以鹽酸小檗堿-磷脂復(fù)合物的復(fù)合率為評價指標(biāo),對復(fù)合過程的超聲時間進行篩選,篩選結(jié)果如圖6所示。分析結(jié)果表明,當(dāng)超聲復(fù)合時間為15 min時,復(fù)合率達到80%左右;超聲時間為30 min時,復(fù)合率達到90%;之后,隨著超聲時間增加,未見復(fù)合率進一步顯著增加。
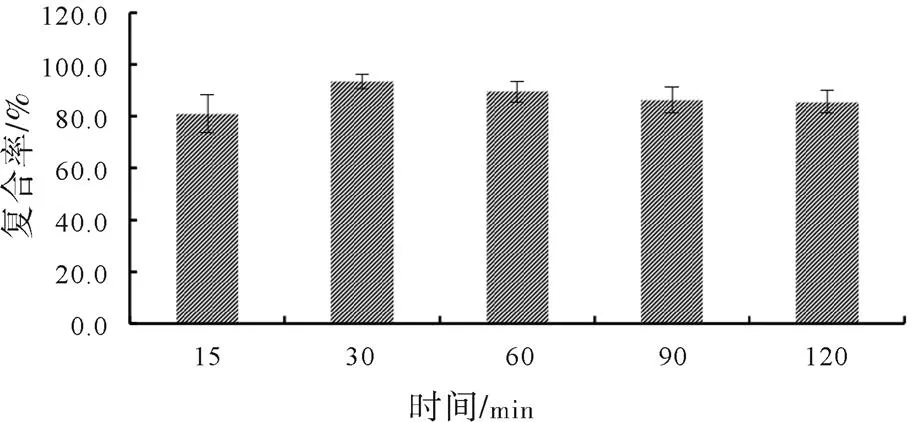
圖6 超聲復(fù)合時間的單因素篩選結(jié)果
3.3 正交設(shè)計試驗
通過3因素3水平(L9,34)正交設(shè)計,以復(fù)合率作為評價指標(biāo),對溶劑系統(tǒng)pH值(A)、鹽酸小檗堿與磷脂的投料比例(B)、超聲時間(C)進行了正交篩選,得出制備鹽酸小檗堿-磷脂復(fù)合物的最佳處方,正交分析結(jié)果如表3所示。
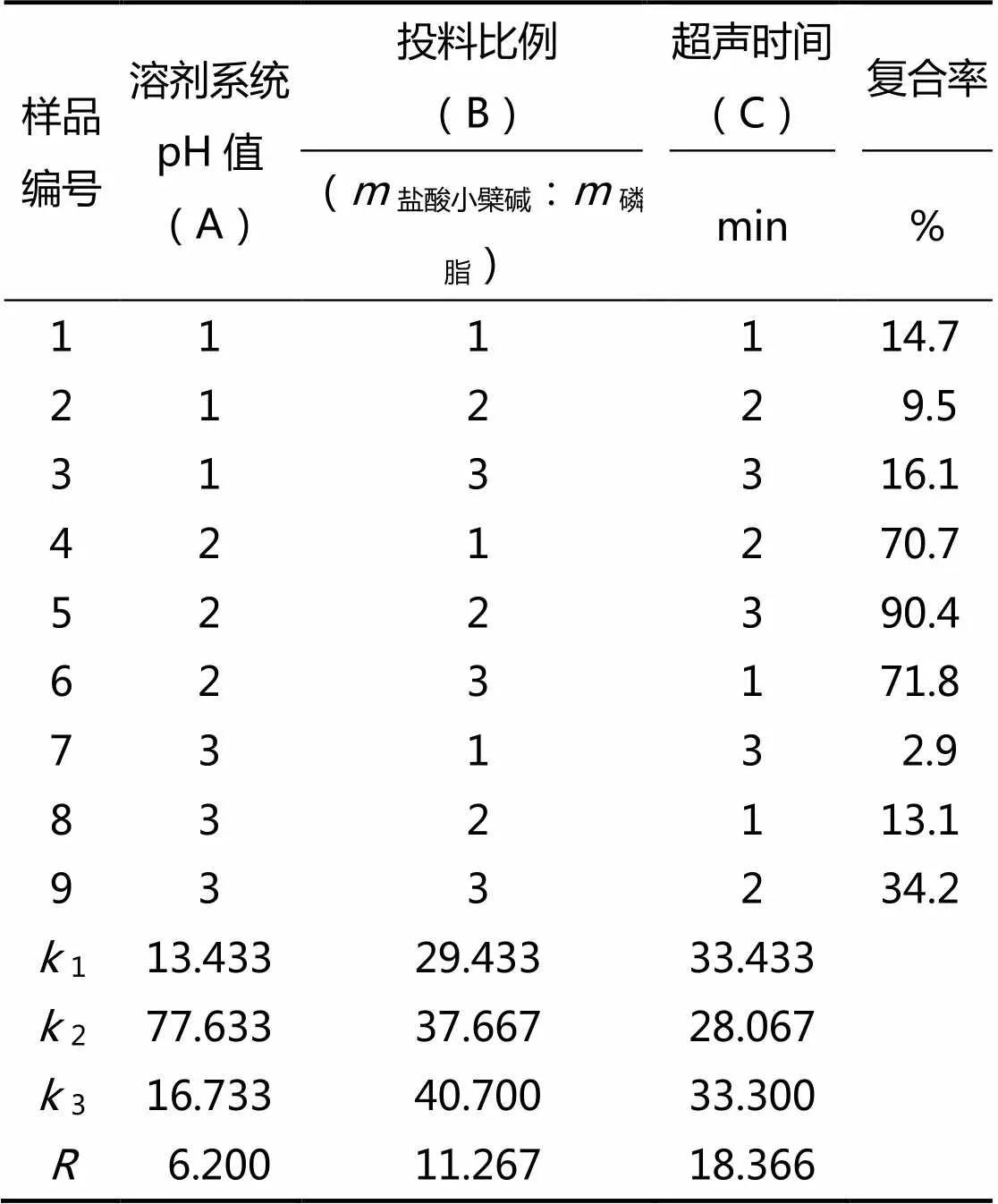
表3 處方篩選的正交試驗結(jié)果
分析結(jié)果表明,在影響該磷脂復(fù)合物復(fù)合率的3大因素中,超聲時間與投料比例對復(fù)合率的影響較大,其次是溶劑系統(tǒng)的pH值。通過正交極差分析得出最優(yōu)的處方為A2B3C1,即溶劑系統(tǒng)的pH值為6.0、鹽酸小檗堿與磷脂的質(zhì)量比例為1∶15、超聲時間為15 min。
3.4 優(yōu)化處方下磷脂復(fù)合物的穩(wěn)定性評價
按照最優(yōu)處方條件制備了3批鹽酸小檗堿-磷脂復(fù)合物,并且對其放置穩(wěn)定性進行了初步性評價表征,結(jié)果如表4所示。考察結(jié)果表明,優(yōu)選處方下,所制備的3批鹽酸小檗堿-磷脂復(fù)合物的復(fù)合率為(91.3±2.38)%,外觀呈黃色顆粒狀。將上述3批復(fù)合物置于4 ℃條件下進行放置穩(wěn)定性評價,結(jié)果表明優(yōu)選處方下鹽酸小檗堿-磷脂復(fù)合物放置3個月以內(nèi)的穩(wěn)定性良好,復(fù)合率以及鹽酸小檗堿含量均未發(fā)生顯著變化。
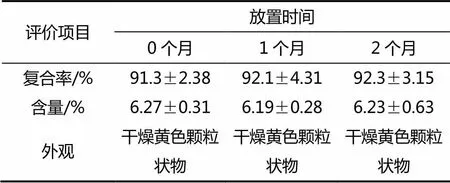
表4 鹽酸小檗堿-磷脂復(fù)合物的放置穩(wěn)定性評價(4 ℃)
4 討論
從本質(zhì)上看,磷脂復(fù)合物是以磷脂作為分散載體(或者分散介質(zhì)),通過適宜的方法制備的一類固體分散體,其中,活性藥物以無定形的狀態(tài)均勻分散在分散載體中,能夠顯著改善難溶性藥物的溶解性、溶解速度以及生物利用度[12-13]。根據(jù)藥物的性質(zhì),可通過多種方法來制備磷脂復(fù)合物,包括冷凍干燥法、熔融法等[14-15]。本研究通過篩選適宜的復(fù)合溶劑系統(tǒng),且聯(lián)合使用超聲及旋轉(zhuǎn)蒸發(fā)法,與已有研究相比,大大縮短了制備時間,同時提高了復(fù)合效率,表現(xiàn)出較大的優(yōu)勢。
研究發(fā)現(xiàn)在鹽酸小檗堿-磷脂復(fù)合物成型性的限制因素中,溶劑的pH值具有一定影響。事實上,在制備過程中發(fā)現(xiàn):如果溶劑的pH值偏離6.0~7.0時,鹽酸小檗堿-磷脂復(fù)合物基本不能復(fù)合成型,大量的鹽酸小檗堿沉淀在底部,無法與磷脂溶液進行復(fù)合。這可能是由于改變pH值影響了鹽酸小檗堿在有機溶劑中的溶解性,導(dǎo)致其無法在均一的溶液中以分子狀態(tài)與磷脂分子發(fā)生復(fù)合作用。因此,提示在后續(xù)的研究中,有必要針對難溶性藥物的溶解狀態(tài)與復(fù)合率的關(guān)系進行相關(guān)研究。
本研究中,通過系統(tǒng)的處方篩選改良了鹽酸小檗堿-磷脂復(fù)合物的處方工藝,縮短了復(fù)合時間的同時提高了復(fù)合效率,有望為其進一步應(yīng)用提供理論基礎(chǔ),具有很大的發(fā)展空間以及實際應(yīng)用價值,對于中藥新劑型的構(gòu)建和改進也具有一定的指導(dǎo)意義。
[1] 李波,朱維良,陳凱先. 小檗堿及其衍生物的研究進展[J].藥學(xué)學(xué)報,2008,43(8):773-787.
[2] 張磊,王京,聶晶,等. 小檗堿衍生物的活性研究進展[J].中國醫(yī)藥指南,2015(12):32-34.
[3] 鐘慈平,騫宇,舒暢,等. 小檗堿及其衍生物抑菌作用研究進展[J]. 食品科學(xué),2013,34(7):321-325.
[4] 王成艷,胡興亞. 小檗堿及其衍生物抗白血病活性的研究進展[J]. 解剖學(xué)研究,2016(3):203-205.
[5] 金鑫,宋霞,曹永兵,等. 小檗堿的結(jié)構(gòu)改造及其藥理活性的研究進展[J]. 藥學(xué)實踐雜志,2017,32(3):171-175.
[6] 李楠,馮玲玲,蔣學(xué)華,等. 黃芩苷磷脂復(fù)合物大鼠在體胃腸道吸收研究[J]. 中國藥學(xué)雜志,2016,51(12):994-998.
[7] 王夢姝,呂曉玲,趙煥焦,等. 黑米花色苷磷脂復(fù)合物的制備及生物利用度[J]. 食品科技,2017(5):242-245.
[8] 鄭林. 磷脂復(fù)合物對中藥制劑口服吸收的影響[J]. 中國抗生素雜志,2015,40(6):468-473.
[9] 左巨波,尚京川. 鹽酸小檗堿磷脂復(fù)合物的制備和理化性質(zhì)研究[J]. 中國新藥雜志,200,18(14):1372-1376.
[10] 華玉鈴,賀祝英,張建玲,等. 中藥軟膏劑制備方法的研究進展[J]. 貴陽中醫(yī)學(xué)院學(xué)報,2008,30(2):66-69.
[11] 崔曉鴿,曹伶俐,侯佳威,等. 白楊素磷脂復(fù)合物的制備及其藥動學(xué)行為[J]. 中成藥,2017,39(5):934-939.
[12] 宋婷,宋丹,管海燕,等. 松蘿酸磷脂復(fù)合物在大鼠體內(nèi)的藥動學(xué)及組織分布研究[J]. 中草藥,2018,49(6):1358-1364.
[13] Shi C,Tong Q,F(xiàn)ang J,et al. Preparation, characterization and in vivo studies of amorphous solid dispersion of berberine with hydrogenated phosphatidylcholine[J]. European Journal of Pharmaceutical Sciences,2015,74:11-17.
[14] Cui F,Shi K,Zhang L Q,et al. Biodegradable nanoparticles loaded with insulin—phospholipid complex for oral delivery:Preparation,in vitro characterization and in vivo evaluation[J]. Journal of Controlled Release,2006,114(2):242-250.
[15] 余鳳瑋,聶績,袁野. 穿心蓮內(nèi)酯磷脂復(fù)合物的制備及表征[J]. 中國藥房,2016,27(10):1409-1411.
Formulation optimization and research on berberine hydrochloride phospholipid complex
WANG Qinga, LIU Li-yaoa, b, Corresponding Author, WANG Qing-kuib
(Tianjin Agricultural University, a. Department of Biopharmaceuticals, College of Basic Science; b. Tianjin Key Lab of Aqua-Ecology and Aquaculture, Tianjin 300384, China)
Berberine hydrochloride is effective medicine for gastrointestinal bacterial infection treatment clinically. However, as for its poor lipophilicity, oral bioavailability of beriberine hydrochloride is relatively low. This study was designed to increase the lipophilicity of berberine hydrochloride using phospholipid complexation technology. In this study, associative efficiency() was set as screening parameter and optimal formulation of berberine hydrochloride phospholipid complex was obtained using solvent evaporation method based on single factor screening and orthogonal design. Under the selected formulation,of berberine hydrochloride phospholipid complex could reach 90% and storage stability remained in 6 months under 4 ℃, which provided the basis for practical application.
berberine hydrochloride; phospholipid complex; orthogonal screening; formulation optimization
R944.9
A
1008-5394(2019)02-0064-05
10.19640/j.cnki.jtau.2019.02.015
2019-01-03
天津市教委科研計劃項目(2017KJ181);天津市水產(chǎn)產(chǎn)業(yè)技術(shù)體系創(chuàng)新團隊項目(ITTFRS2017004);國家自然科學(xué)基金面上項目(31170442)
王慶(1996-),男,本科在讀,主要從事藥物制劑方面的研究。E-mail:1051123854@qq.com。
劉黎瑤(1988-),女,講師,博士,主要從事藥物制劑方面的研究。E-mail:llymeilin@163.com。
責(zé)任編輯:張愛婷
