HPLC同時測定炙黃芪配方顆粒特征圖譜及毛蕊異黃酮葡萄糖苷含量△
2019-07-13 03:16:58姚娜黃燕明李雪銀陳桂生
中國現代中藥 2019年6期
姚娜,黃燕明,李雪銀,陳桂生
1.廣州市香雪制藥股份有限公司,廣東 廣州 510663;2.寧夏隆德縣六盤山中藥資源開發有限公司,寧夏 固原 756300
炙黃芪配方顆粒是以炙黃芪飲片為原料,經水提、濃縮、干燥、制粒、包裝等工藝制備而成。原料炙黃芪為豆科植物蒙古黃芪Astragalusmembranaceus(Fisch.)Bge.var.mongholicus(Bge.)Hsiao或膜莢黃芪Astragalusmembranaceus(Fisch.)Bge.的干燥根炮制加工品。其具有益氣補中,臨床上用于氣虛乏力,食少便溏[1]。黃芪中的主要化學成分研究及相關的藥理作用已有報道[2-4],其中毛蕊異黃酮葡萄糖苷是主要的有效成分之一。黃芪經炮制后,毛蕊異黃酮葡萄糖苷含量有明顯變化[5-7]。目前國內尚無關于HPLC同步測定炙黃芪配方顆粒特征圖譜和毛蕊異黃酮葡萄糖苷含量的文獻報道,國家也尚未出臺該中藥配方顆粒的質量標準。為了有效控制和評價炙黃芪配方顆粒的質量,本研究采用HPLC建立了同步測定炙黃芪配方顆粒的特征圖譜并對指標成分毛蕊異黃酮葡萄糖苷進行定量檢測的方法[8]。
1 材料
1.1 儀器
十萬分之一電子分析天平(MS205DU,瑞士Mettler toledo公司);超純水器(Simplicity,美國密理博Millipore公司);數控超聲波清洗器(KQ500DE,昆山市超聲儀器有限公司);Ultimate 3000 DGLC高效液相色譜儀;Agilent 1200高效液相色譜儀。
1.2 試藥
蒙古黃芪對照藥材(中國食品藥品檢定研究院,批號:120974-201311,規格:1 g/瓶);膜莢黃芪對照藥材(中國食品藥品檢定研究院,批號:121462-201304,規格3 g/瓶);毛蕊異黃酮葡萄糖苷(中國食品藥品檢定研究院,批號:111920-201505,純度:97.1%);乙腈、甲酸均為色譜級;甲醇為分析純。
炙黃芪配方顆粒10批來自廣州市香雪制藥股份有限公司,批號依次為S1~S10;麥芽糊精來自湖州展望藥業有限公司。
2 方法與結果
2.1 色譜條件與系統適用性試驗
以十八烷基硅烷鍵合硅膠為填充劑(250 mm×4.6 mm,5 μm);以乙腈為流動相A,以0.2%甲酸溶液為流動相B,梯度洗脫方式0~40 min,10%~50%A,90%~50%B;檢測波長為254 nm;柱溫25 ℃;進樣量為10 μL。理論塔板數按毛蕊異黃酮葡萄糖苷峰計算應不低于3000。
2.2 對照品溶液的制備
取毛蕊異黃酮葡萄糖苷對照品適量,精密稱定,加甲醇制成每1 mL含50 μg的溶液,即得。
2.3 供試品溶液的制備
取炙黃芪配方顆粒約1 g,研細,精密稱定,精密加入甲醇50 mL,稱定質量,加熱回流2 h,放冷,再稱定質量,用甲醇補足減失的質量,搖勻,濾過,精密量取續濾液25 mL,回收溶劑至干,殘渣加甲醇溶解,轉移至5 mL量瓶中,加甲醇至刻度,搖勻,即得。
2.4 測定法
分別精密吸取對照品溶液與供試品溶液各10 μL,注入液相色譜儀,測定,記錄色譜圖。
2.5 10批樣品的檢測結果
分別取10批炙黃芪配方顆粒,按2.3項下方法制備10份供試品溶液,按2.1項下的色譜條件測定,得到10批炙黃芪配方顆粒的毛蕊異黃酮葡萄糖苷的質量分數分別為0.317 2、0.309 1、0.317 3、0.311 2、0.318 5、0.313 3、0.678 8、0.691 3、0.683 4、0.687 8 mg·g-1,平均質量分數為0.462 8 mg·g-1;得到10批炙黃芪配方顆粒的色譜疊加圖,見圖1,通過對10批的色譜圖進行分析,有4個色譜峰能穩定重現,故確立了4個特征峰,其中1號峰的保留時間及紫外光譜與毛蕊異黃酮葡萄糖苷對照品吻合,故供試品特征圖譜中1號峰鑒定為毛蕊異黃酮葡萄糖苷,相對保留時間的規定值為:1.00(峰1)、1.46(峰2)、1.75(峰3)、2.38(峰4)。
2.6 特征圖譜方法學考察
2.6.1 專屬性試驗 取炙黃芪配方顆粒、麥芽糊精、蒙古黃芪對照藥材及膜莢黃芪對照藥材分別按2.3項下方法制備,按2.1項下的色譜條件測定,見圖2。結果表明,4個共有色譜峰的鑒定均不受溶劑及輔料等因素干擾,具有良好的專屬性。
2.6.2 精密度試驗 取同一批炙黃芪配方顆粒供試品溶液,按2.1項下的色譜條件于高效液相色譜儀上連續進樣6次,記錄色譜圖。以1號峰毛蕊異黃酮葡萄糖苷峰為參照,對各圖譜的特征峰的相對保留時間進行計算,結果各圖譜特征峰相對保留時間的RSD值為0%~0.07%,各主要色譜峰的相對保留時間無明顯變化,表明儀器精密度良好。
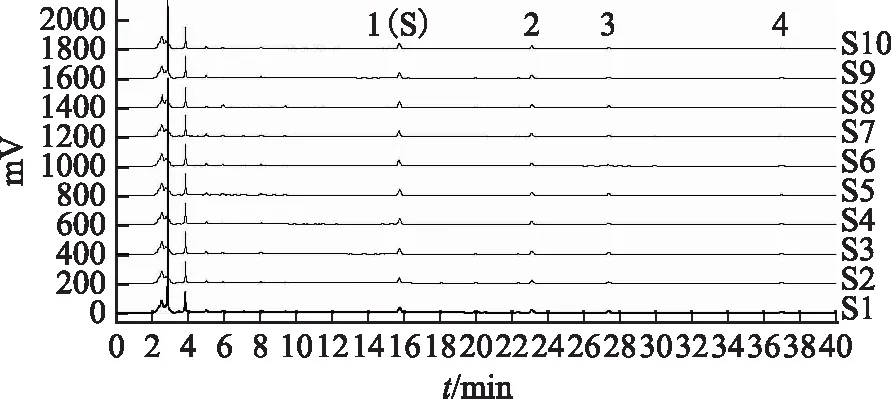
注:S1.201503001批次;S2.201503002批次;S3.201503003批次;S4.201503004批次;S5.201503005批次;S6.201503006批次;S7.201503007批次;S8.201503008批次;S9.201503009批次;S10.201503010批次。圖1 10批炙黃芪配方顆粒HPLC特征圖譜疊加圖
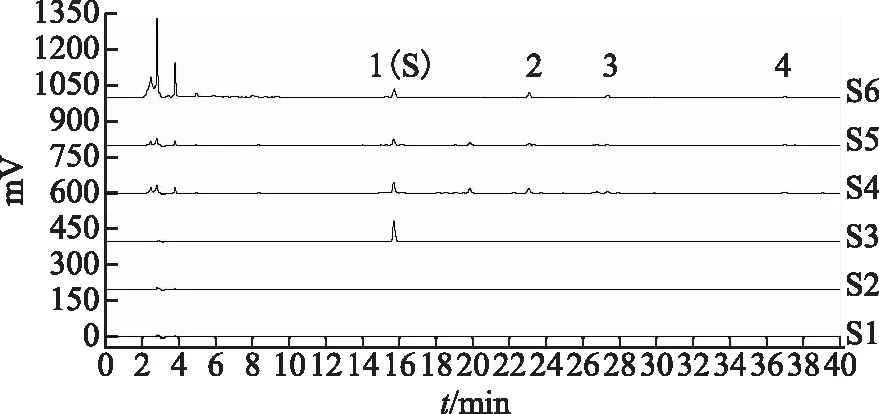
注:S1.空白溶劑;S2.陰性對照;S3.毛蕊異黃酮葡萄糖苷對照品;S4.蒙古黃芪對照藥材;S5.膜莢黃芪對照藥材;S6.炙黃芪配方顆粒。圖2 炙黃芪配方顆粒特征圖譜專屬性試驗
2.6.3 重復性試驗 取同一批炙黃芪配方顆粒樣品,平行6份,按2.3項下方法制備,按2.1項下的色譜條件分別進樣,記錄色譜圖。以1號峰毛蕊異黃酮葡萄糖苷峰為參照,對各圖譜的特征峰的相對保留時間進行計算,結果各圖譜特征峰相對保留時間的RSD值為0%~0.07%,各主要色譜峰的相對保留時間無明顯變化,表明方法的重復性良好。
2.6.4 穩定性試驗 取同一批炙黃芪配方顆粒供試品溶液,按2.1項下色譜條件,分別在0、2、4、6、8、12、24、48 h進樣,記錄色譜圖。以1號峰毛蕊異黃酮葡萄糖苷峰為參照,對各圖譜的特征峰的相對保留時間進行計算,結果各圖譜特征峰相對保留時間的RSD值為0%~0.07%,各主要色譜峰的相對保留時間無明顯變化,表明供試品溶液在48 h內穩定性良好。
2.6.5 中間精密度試驗 分別考察不同分析人員、不同日期、不同設備對精密度的影響,結果不同影響因素下各圖譜特征峰相對保留時間的RAD值在0%~0.63%,各主要色譜峰的相對保留時間無明顯變化,表明該方法精密度良好,隨機變動因素不影響該方法精密度。
2.6.6 耐用性試驗 取同一批炙黃芪配方顆粒供試品,分別使用Agilent Zorbax Eclipse Plus C18(250 mm×4.6 mm,5 μm)、Elite Hypersil ODS2(250 mm×4.6 mm,5 μm)、Welch Ultimate XB-C18(250 mm×4.6 mm,5 μm)3種型號的色譜柱,測定炙黃芪配方顆粒的特征圖譜,記錄色譜圖。
以1號峰毛蕊異黃酮葡萄糖苷峰為參照,各圖譜特征峰相對保留時間的RSD值小于2.84%,各主要色譜峰的相對保留時間無明顯變化,表明該方法耐用性良好。
2.7 含量測定方法學考察
2.7.1 線性關系及范圍 取毛蕊異黃酮葡萄糖苷對照品溶液(質量濃度:49.33 μg·mL-1),采用自動進樣器分別進樣0.1、0.5、1、2、5、10、15、20 μL,在260 nm下測定,即毛蕊異黃酮葡萄糖苷進樣量為0.004 9、0.024 7、0.098 7、0.246 6、0.493 3、0.740 0、0.986 6 μg,以毛蕊異黃酮葡萄糖苷峰面積積分值對毛蕊異黃酮葡萄糖苷對照品的進樣量進行回歸分析,其回歸方程為:Y=37.68X-0.016,相關系數r=1;結果表明,毛蕊異黃酮葡萄糖苷在0.004 9~0.986 6 μg,濃度與峰面積呈良好線性關系。
2.7.2 重復性試驗 同2.6.3項下進行含量測定,并對所得數據進行處理,RSD為2.88%,結果表明該方法重復性良好。
2.7.3 穩定性試驗 同2.6.4項下測定,對照品溶液和供試品溶液的RSD分別為0.41%、1.05%,表明炙黃芪配方顆粒供試品溶液及毛蕊異黃酮葡萄糖苷對照品溶液在48 h內穩定性良好。
2.7.4 加樣回收率試驗 精密稱取已測毛蕊異黃酮葡萄糖苷含量的炙黃芪配方顆粒適量,平行9份,分別精密加入低、中、高濃度的毛蕊異黃酮葡萄糖苷對照品,按供試品項下操作,依法測定,平均回收率為96.26%,RSD為2.80%,結果表明該方法回收率良好。
2.7.5 中間精密度試驗 同2.6.5項下測定,毛蕊異黃酮葡萄糖苷含量的RAD在0.11%~0.59%,表明該方法中間精密度良好。
2.7.6 耐用性試驗 同2.6.6項下測定,毛蕊異黃酮葡萄糖苷含量的RSD為0.56%,表明該方法耐用性良好。
3 討論
特征圖譜方法研究中,參考文獻[9],進行流動相梯度洗脫時發現,炙黃芪配方顆粒主要色譜峰集中在乙腈-0.2%甲酸溶液10∶90~50∶50,且毛蕊異黃酮葡萄糖苷峰出峰時間過早,故優化流動相比例。
在毛蕊異黃酮葡萄糖苷含量測定中,對其定量下限進行研究。取毛蕊異黃酮葡萄糖苷對照品溶液(質量濃度:49.33 μg·mL-1)適量,進樣0.1 μL,依法測定。信噪比約為20∶1時,對應毛蕊異黃酮葡萄糖苷的量為0.004 9 μg,即本含量測定方法的定量限0.004 9 μg。
對蒙古黃芪和膜莢黃芪的特征圖譜進行對比發現,蒙古黃芪與膜莢黃芪中均含4個特征峰,未見明顯區別。
綜上所述,實驗中建立的采用HPLC同步檢測炙黃芪配方顆粒毛蕊異黃酮葡萄糖苷含量與特征圖譜方法穩定性、重復性好,為炙黃芪配方顆粒質量控制和評價提供了參考。
