三七總皂苷對羧酸酯酶體外活性的影響△
2019-07-13 03:47:10齊琪王彥莫雨佳羅菊元喻祥龍陸洋杜守穎
中國現代中藥 2019年6期
齊琪,王彥,莫雨佳,羅菊元,喻祥龍,陸洋,杜守穎
北京中醫藥大學 中藥學院,北京 102488
羧酸酯酶(Carboxylesterases,CESs),屬于α/β水解酶超家族成員,是體內重要的Ⅰ相藥物代謝酶,在不同種屬動物體內普遍存在。人體內主要為羧酸酯酶1(hCE1)和羧酸酯酶2(hCE2),這兩者氨基酸序列的同源性為48%,在組織分布、底物特異性等方面存在較大差異[1-2],hCE1在肝臟中高表達[3],hCE2在小腸和結腸中高表達[4]。人類與常用實驗動物在hCE1和hCE2的分布上也存在顯著的種屬差異。羧酸酯酶除參與小分子脂肪酸和膽固醇的運輸代謝外[5],還會直接影響藥物的首過代謝(腸、肝),因藥物導致相關羧酸酯酶的表達與活性變化是藥-藥相互作用的重要原因之一。如抗癌藥伊立替康和卡培他濱等被設計成無活性的酯類前體藥,經羧酸酯酶水解后起效,當聯用白花前胡丁素(hCE1強抑制劑)[6]或五味子甲素(hCE2強抑制劑)[7]時,可使抗癌效果降低。又如抗血栓藥氯吡格雷大部分在肝臟中經CESs代謝失活,剩下約15%經CYP酶系轉化為活性代謝產物,CESs代謝能力的變化會影響氯吡格雷在體內的分布代謝,進而影響活性代謝產物的產生和藥效的發揮[8]。
三七是一種散瘀止血常用中藥,有很好的抗缺血損傷和抗腦缺血再灌注損傷活性[9],在心腦血管疾病中,常與化學藥共用。如在阿司匹林與三七總皂苷相關制劑(血塞通、血栓通等)合用治療腦卒中,兩者聯用后抗血小板凝集作用強于阿司匹林或三七總皂苷單用[10]。體內參與阿司匹林代謝的酯酶統稱為阿司匹林酯酶,它主要作用于酯鍵的羧酸酯水解酶。阿司匹林原型藥物可發揮藥效作用,被水解為水楊酸后無抗血小板聚集作用。本課題組前期研究發現,三七總皂苷可降低阿司匹林的體內代謝,這可能是聯用增效的原因之一。為進一步明確三七總皂苷對阿司匹林的作用機制,本實驗在體外肝微粒體水平下對三七總皂苷抑制羧酸酯酶活性的能力進行探究,為三七聯用羧酸酯酶底物提供合理的科學依據。
1 材料與儀器
1.1 材料
熒光素二乙酸酯(FDA)和熒光素(F)(上海阿拉丁生化科技股份有限公司,純度>97%),2-(2-苯甲酰基-3-甲氧基苯基)-苯并噻唑(BMBT)和其水解產物(HMBT)(大連化學物理研究所,純度>98%),雙(4-硝基苯基)磷酸酯(BNPP)(東京化成工業株式會社,純度>98%),三七總皂苷原粉(PNS)(云南三七科技有限公司,批號:110870-201603),小鼠肝微粒體(MLM)(美國Corning Gentest公司),二甲基亞砜(DMSO)(北京化工廠,分析純),乙腈[賽默飛世爾科技(中國)有限公司,色譜純],磷酸鹽緩沖液(PBS)(美國Corning公司,1×,pH 7.4)。
1.2 儀器
BS224型電子天平(德國賽多利斯公司),微量移液器(德國艾本德公司),307057型空氣浴振蕩搖床(江蘇太倉豪誠實驗儀器制造有限公司),5427型高速離心機(德國艾本德公司),SpectraMax i3x型多功能酶標儀(美國Molecuar Devices LLC公司)。
2 方法與結果
2.1 方法
在200 μL的反應體系中,含有PBS緩沖液(100 mmol·L-1,pH 7.4)及適量的小鼠肝微粒體,渦旋混合均勻后,于37 ℃恒溫空氣浴共同振蕩孵育活化10 min,向體系中分別添加適量的熒光探針BMBT(hCE1特異性底物)和FDA(hCE2特異性底物)起始反應,37 ℃振蕩孵育20 min后,加入等體積冰乙腈渦旋終止反應,8000 r·min-1離心3 min去除蛋白。移取200 μL上清液至96孔板,于熒光酶標儀測量熒光強度(FDA水解產物F激發波長為480 nm,發射波長為520 nm;BMBT水解產物HBMT激發波長為304 nm,發射波長為488 nm)。
2.1.1 反應條件優化
2.1.1.1 底物濃度優化 肝微粒體濃度保持2 μg·mL-1(BMBT),0.2 μg·mL-1(FDA),將底物濃度設為1、2、5、10、20、30 μmol·L-1;酶活化后加入探針底物,反應2 min,每15 s測定熒光強度,時間和熒光強度線性擬合得到的斜率即為反應速率,將底物濃度和反應速率代入米氏方程進行非線性擬合得到動力學常數Km。
2.1.1.2 肝微粒體濃度優化 采用接近Km的底物濃度,將肝微粒體質量濃度設為0.25、0.5、1、2、3 μg·mL-1(底物為BMBT),0.1、0.15、0.2、0.3、0.5 μg·mL-1(底物為FDA);按2.1方法測量熒光強度,繪制酶濃度-熒光強度曲線。
2.1.1.3 DMSO用量優化 采用優化好的底物濃度和肝微粒體濃度,將溶解探針底物的DMSO用量設為0%、1%、2%、3%(V/V);按2.1方法測量熒光強度,按公式(1)計算羧酸酯酶殘余活性。

(1)
2.1.1.4 乙腈用量優化 采用優化好的底物濃度和肝微粒體濃度,在活化時分別加入0%、50%、75%、100%(V/V)的冰乙腈,再按2.1方法離心后向反應體系中補加冰乙腈,使冰乙腈終體積均為200 μL,取200 μL上清液至96孔板,于熒光酶標儀測量熒光強度,計算羧酸酯酶殘余活性。
2.1.2 三七總皂苷對羧酸酯酶的抑制作用 采用優化好的酶促反應條件,將三七總皂苷質量濃度設為1、10、100 μg·mL-1,與酶共孵育活化,按2.1方法測量熒光強度,并公式(2)計算對羧酸酯酶活性的抑制率。陰性對照(等體積PBS代替三七總皂苷)和陽性對照(等體積羧酸酯酶陽性抑制劑BNPP,終濃度100 μmol·L-1代替三七總皂苷)在相同條件下反應。選擇抑制效果較為明顯的羧酸酯酶亞型,將三七總皂苷質量濃度設為1、5、10、20、50、100、300、500、700 μg·mL-1,計算三七總皂苷對羧酸酯酶活性的抑制率,并計算IC50值。
抑制率=1-殘余活性
(2)
2.1.3 抑制類型的判斷 選擇5組三七總皂苷質量濃度,采用優化好的酶促反應條件,將三七總皂苷與肝微粒體共孵育活化,按“底物濃度優化”方法測量動力學常數(Km)和最大反應速率(Vmax),轉化為雙倒數方程,判斷抑制類型。
2.1.4 體外-體內外推 通過抑制劑濃度和相對應的雙倒數方程的斜率計算求得抑制動力學參數Ki;體外-體內外推使用的方程為:AUCi/AUC=1+C抑制劑invivo/Ki,若C抑制劑invivo/Ki<0.1,無藥物相互作用可能性;0.1
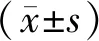
2.2 結果
2.2.1 最適反應條件
2.2.1.1 最適底物濃度 通過米氏方程非線性擬合(如圖1),當底物為BMBT時,獲得的動力學常數Km=(12.79±2.427)μmol·L-1,BMBT最適濃度為13 μmol·L-1;當底物為FDA時,獲得的動力學常數Km=(7.866±1.367)μmol·L-1,FDA最適濃度為8 μmol·L-1。

注:A.BMBT;B.FDA。圖1 小鼠肝微粒體中BMBT(A)/FDA(B)水解產物的動力學行為
2.2.1.2 最適肝微粒體濃度 為保證實驗結果準確,底物水解率不得高于20%。當底物為BMBT時,熒光強度(Y)與肝微粒體濃度(X)回歸方程表示為Y=380 120X-34 674(r=0.996 0),BMBT的最適肝微粒體濃度為2 μg·mL-1;當底物為FDA時,回歸方程表示為Y=6.40×108X-1.10×107(r=0.996 5),FDA的最適肝微粒體濃度為0.2 μg·mL-1。見圖2。

注:A.BMBT;B.FDA。圖2 BMBT(A)/FDA(B)水解產物熒光強度隨肝微粒體濃度的線性變化
2.2.1.3 有機溶劑用量 結果表明,當體系中存在0%~3% DMSO時,hCE1和hCE2的活性沒有顯著變化;體系中加入一半體積以上的冰乙腈可以終止酶促反應,最終確定加入200 μL冰乙腈。
2.2.2 三七總皂苷對羧酸酯酶的抑制作用 如圖3所示,當質量濃度為100 μg·mL-1時,三七總皂苷對hCE1的抑制率僅為27%;而三七總皂苷質量濃度為10 μg·mL-1時,對hCE2的抑制率達到42.1%,當質量濃度為100 μg·mL-1時,抑制率為63.5%,抑制效果較為明顯(見圖4),進一步細化三七總皂苷濃度進行抑制率實驗,求得三七總皂苷對hCE2的半數抑制率IC50為17.05 μg·mL-1。

注:A.hCE1;B.hCE2;NC代表陰性對照;PC代表陽性對照。圖3 三七總皂苷對hCE1(A)/hCE2(B)活性抑制效果的初篩

圖4 三七總皂苷對hCE2活性的抑制作用
2.2.3 抑制類型 通過雙倒數方程對實驗結果進行分析,所得的雙倒數擬合直線相交于X軸(如圖5),說明三七總皂苷對hCE2的抑制類型為非競爭型抑制;將各斜率和對應的三七總皂苷濃度進行方程擬合,所得函數為Y=0.003 7X+0.29,進一步計算求得三七總皂苷對hCE2的Ki為77.53 μg·mL-1。
2.2.4 體內藥-藥相互作用的發生可能性 大鼠在灌胃三七總皂苷(600 mg·kg-1)后,采用整合藥代動力學的方法,擬合出三七總皂苷的Cmax為18.473 μg·mL-1[12],將此濃度(C抑制劑)代入體外-體內外推方程,求得C抑制劑/Ki值為0.24,這表明由于抑制hCE2活性所造成的藥物生物利用度比未服用三七總皂苷時增大了0.24倍。

注:A.雙倒數方程;B.抑制動力學參數方程。圖5 三七總皂苷對hCE2活性抑制的雙倒數方程和抑制動力學參數方程
3 討論
本研究證明,三七總皂苷可較明顯地直接抑制hCE2的水解活性,且通過體外-體內外推方法證明,三七總皂苷在與經hCE2代謝的藥物一起服用時,需要警惕藥-藥相互作用的發生。在體外水平下,三七總皂苷對hCE1的活性表現出微弱的直接抑制作用,但是三七總皂苷對hCE1的活性是否有間接抑制作用還需要進一步考察:由于選擇肝微粒體作為酶源,并不存在羧酸酯酶的表達翻譯過程,但三七總皂苷還有可能通過影響hCE1的轉錄翻譯環節,從而抑制其水解活性。
羧酸酯酶的催化中心由一個包含絲氨酸催化三聯體的中央催化位點組成,絲氨酸殘基很容易與含羰基分子反應形成中間體,若中間體的碳碳雙鍵無法斷裂,則會抑制催化活性。具有兩個相鄰羰基結構1,2-二酮類分子大多可強烈抑制羧酸酯酶的活性[13],而三七總皂苷中所含有的5種皂苷(三七皂苷R1、人參皂苷Rg1、人參皂苷Re、人參皂苷Rb1、人參皂苷Rd)均不具有這種可以和活性催化中心結合的相鄰羰基結構,說明三七總皂苷對羧酸酯酶的抑制類型不可能是競爭型抑制,本實驗進一步研究發現三七總皂苷對hCE2的抑制類型為非競爭型抑制,說明三七總皂苷與hCE2的非活性部位相結合,形成抑制物-酶的絡合物后進一步再與底物結合;或是hCE2與底物結合成底物-酶絡合物后,其中有部分再與三七總皂苷結合。這兩種情況所形成的中間絡合物都不能直接生成水解產物,從而導致了酶催化反應速率的降低。
三七作為一種中長期服用的中藥,在腸道中對羧酸酯酶的抑制效果可能是累積作用。因此,長期服用三七總皂苷,同時使用羧酸酯酶底物藥物時應該建議進行體內藥物水平的監測,必要時可調整用藥量,以確保臨床安全有效。
