林奇綜合征相關的卵巢癌一例報告
2019-07-06 10:30:47趙劉碧琦徐子力王芳于倩倩劉宗諭范麗梅
新醫學 2019年8期
趙劉碧琦 徐子力 王芳 于倩倩 劉宗諭 范麗梅

【摘要】林奇綜合征(LS),又稱為遺傳性非息肉性結直腸癌,屬于常染色體顯性遺傳性疾病,由 DNA錯配修復(MMR)基因MLH1、MSH2、 MSH6 和(或)PMS2的胚系突變引起,MMR基因突變也增加了卵巢癌的患病率。該文報道了1例LS相關卵巢癌患者的診療經過。該例患者輔助檢查提示卵巢癌可能性大,術后免疫組織化學染色檢查結果提示可能合并LS,最終行基因檢查確診。LS為遺傳性腫瘤綜合征,其發病可同時或異時發生不同部位惡性腫瘤,臨床醫師應提高對此病的認識,增強早期篩查意識,提升診治水平。
【關鍵詞】林奇綜合征;卵巢癌;遺傳性疾病
【Abstract】Lynch syndrome, also known as hereditary non-polyposis colorectal cancer (HNPCC), is an autosomal dominant inherited cancer susceptibility syndrome. It is caused by the mutations in the DNA mismatch repair (MMR) genes, such as MLH1, MSH2, MSH6 and (or) PMS2. MMR gene mutation can also increase the risk of ovarian cancer. In this article, the diagnosis and treatment of one case of Lynch syndrome-associated ovarian cancer were reported. Auxiliary examination of this patient indicated a high possibility of ovarian cancer. Postoperative immunohistochemical staining suggested that it might be complicated with Lynch syndrome, which was finally confirmed by genetic examination. Lynch syndrome is a hereditary tumor syndrome, and it is prone to occur at the same time or at different time in different parts of malignant tumors. Clinicians should improve the awareness of this disease, enhance the awareness of early screening and improve the level of diagnosis and treatment.
【Key words】Lynch syndrome;Ovarian cancer;Hereditary disease
林奇綜合征(LS)是一種常染色體顯性遺傳性癌癥易感性疾病,DNA錯配修復(MMR)基因MLH1、MSH2、MSH6、PMS2可以糾正 DNA復制過程中產生的錯配,以保持基因組的穩定性。MMR 功能異常或缺失可導致表型突變和微衛星不穩定性 (MSI),從而促進惡性腫瘤的發生[1]。
病例資料
一、病史與體格檢查
患者女,48歲,因腹脹14 d、發現盆腔腫物2 d于2018年9月20日入院。患者在14 d前開始出現腹脹,未在意,平素月經周期規律,末次月經2018年9月11日,因自覺當月月經量大、經期短,于9月19日就診當地醫院并行婦科超聲檢查,結果提示右卵巢囊性腫塊、盆腹腔大量積液,建議就診上級醫院。遂于次日至我院求診。生育史:孕3產1,1996年剖宮產一健康女嬰,自然流產1次,人工流產1次。乙型肝炎史23年,否認家族遺傳病,父親因結腸癌病逝。
體格檢查:生命體征平穩,心、肺未見明顯異常。專科檢查:陰道及宮頸檢查未見明顯異常,子宮正常大,邊界不清,活動度差,右附件區可觸及一直徑約5 cm腫物,形態不規則,質硬,活動度欠佳,左附件區可觸及一直徑約7 cm腫物,形態不規則,質硬,活動度欠佳。
二、實驗室及輔助檢查
入院時血常規:血紅蛋白109 g/L,紅細胞4.0×1012/L,白細胞 5.2×109/L,中性粒細胞0.81,淋巴細胞0.10,單核細胞0.09。糖類抗原199 80.6 kU/L,糖類抗原153 78.4 kU/L,糖類抗原125 > 1 000 kU/L,細胞角蛋白19片段(CYFRA21-1)33.12 μg/L。宮頸薄層液基細胞檢查:送檢標本內偶見有極少量意義不明確的不典型鱗狀上皮細胞(ASCUS),請結合臨床。
婦科陰道彩色多普勒超聲(彩超)示:子宮后壁低回聲3.7 cm×2.9 cm,右附件區5.8 cm×4.5 cm
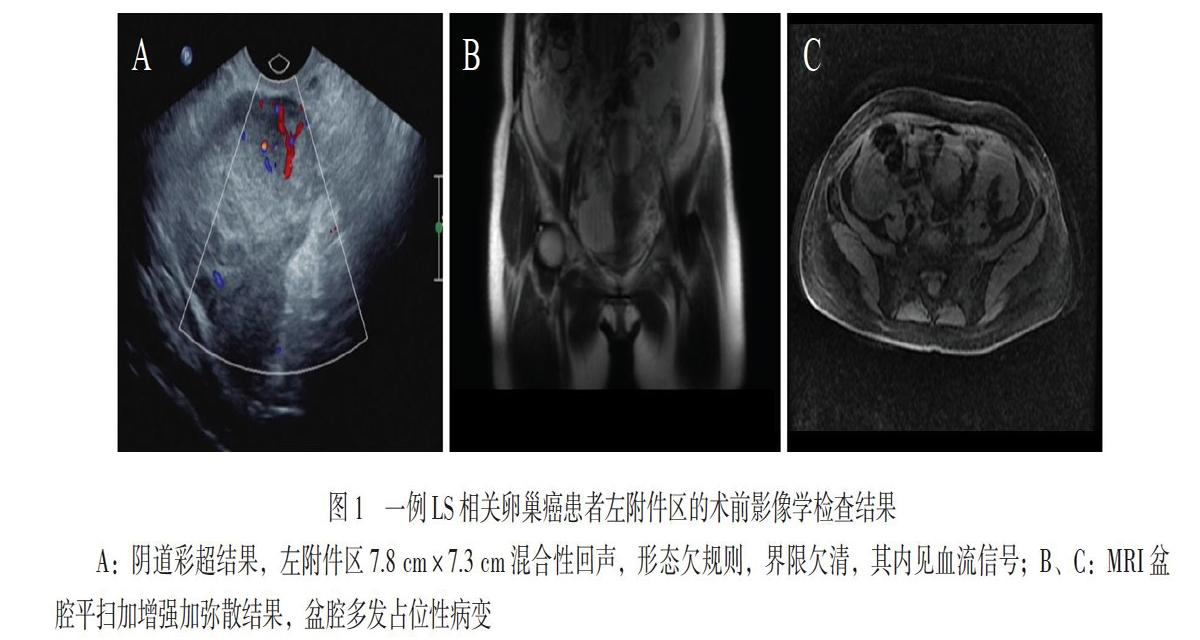
不均質回聲,形態欠規則,界限欠清,其內見血流信號,阻力指數0.79;左附件區7.8 cm×7.3 cm混合性回聲(圖1A),形態欠規則,界限欠清,其內見血流信號;盆腹腔積液5.5 cm。MRI盆腔平掃加增強加彌散檢查示:盆腔多發占位性病變(圖1B、C),考慮來源于卵巢惡性腫瘤性病變可能性大;子宮多發肌瘤;子宮左側壁肌層異常信號;左側髂血管旁及腹股溝區淋巴結顯示;盆腹腔積液;子宮直腸陷窩內結節影,不除外腹膜轉移所致;宮頸囊腫。無痛胃鏡示慢性淺表性胃炎、胃息肉,術后病理回報為(胃底)胃底腺息肉。無痛腸鏡示慢性直腸炎(腸道準備欠滿意,建議擇期復查)。
三、診治過程
予剖腹探查術,術中見大量淡黃色腹水,子宮略增大,雙側卵巢不規則增大,表面凹凸不平,呈菜花樣,質脆,與周圍組織粘連,左側輸卵管增粗,質硬,右側輸卵管大致正常。腸管表面見粟粒樣病灶結節分布,色白,質硬,小腸部分腸管粘連,約60 cm,乙狀結腸表面可見多個直徑> 1 cm病灶結節,直腸表面見約6 cm×8 cm病灶。前腹膜可見多個病灶結節,腹壁可見粟粒樣病灶結節分布,色白,質硬。術中切除雙側附件送檢快速病理回報:(雙側卵巢、左側輸卵管及右側輸卵管系膜)內見腺癌。遂行卵巢癌減滅術、全子宮及雙側附件切除術、腹膜后淋巴結清掃術、大網膜切除術,請外科協同行闌尾切除術和部分小腸、乙狀結腸、直腸切除吻合術。術后病理回報:雙側附件內見癌浸潤,結合免疫組織化學(免疫組化)染色結果支持高級別漿液性癌,脈管內見癌浸潤(因多個部位可見有漿液性癌,請結合臨床進一步確定原發部位,排除腹膜源性漿液性癌)。免疫組化:PMS2(+)、MLH1(+)、MSH2(+)、ER(+)、PR(部分+)、P16(+)、P53(-)、Ki67(陽性率70%)、WT1(+)、CK7(+)、CK20(-)、Calretinin(-)、PAX-8(+),見圖2。左側宮旁、右側漏斗韌帶、乙狀結腸系膜、大網內見癌浸潤,子宮肌壁及宮頸壁內見癌浸潤,脈管見廣泛癌栓,子宮平滑肌瘤,增生型宮內膜。闌尾未見癌,部分小腸腸壁漿膜面見癌浸潤,脈管內見癌栓、切緣未見癌,系膜淋巴結(2/6)見癌轉移,部分乙狀結腸腸壁漿膜面見癌浸潤、切緣未見癌,部分直腸腸壁見癌浸潤、切緣未見癌,系膜淋巴結(3/5)見癌轉移。結合患者罹患卵巢癌、部分乙狀結腸及直腸有癌浸潤,且患者1級親屬患有結腸癌,結合術后病理免疫組化結果,診斷為可疑 LS 突變基因攜帶者。抽取患者外周血,送Novogene公司進行遺傳基因檢測,結果顯示患者 MSH1及PMS2 基因存在突變,確診LS。術后患者未行化學治療及放射治療,隨訪至今患者生存質量尚可,暫未見腫瘤復發。
討論
LS是結直腸癌中最常見的遺傳性腫瘤綜合征,是一種常染色體顯性遺傳病,MMR基因突變可同時或異時發生結直腸癌、胃癌、膽管癌、子宮內膜癌、卵巢癌、輸尿管癌、小腸癌等中的一種或多種,在首發腫瘤確診后,10年內有25%幾率罹患另一種腫瘤,首發腫瘤為卵巢癌的患者,其患有另一種腫瘤的中位時間為5.5年,以子宮內膜癌為首發的患者,其時間為11年,子宮內膜癌和卵巢癌是LSⅡ型綜合征最常見的腸道外表現,發病率高于或等于結直腸癌,可視為LS的“前哨”腫瘤[2-3]。研究表明, LS相關性卵巢癌早期診斷與預后密切相關[4]。因此,LS相關性子宮內膜癌和卵巢癌在婦科及腫瘤領域受到關注。
LS其發病機制是MMR(如MLH1、MSH2、MSH6、PMS2)的種系突變和MSI[5]。微衛星是分布于人類基因組的短重復序列,當MMR功能異常時,重復基因片段可能發生突變,產生“MSI現象”,導致癌基因的激活或抑癌基因的失活,從而誘發癌變。卵巢癌患者MMR突變類型中,最常見是MLH1(38%)和MLH2(47%)。
目前 LS 患者的診斷標準大多依據阿姆斯特丹標準Ⅱ和修訂的 Bethesda 指南[6]。阿姆斯特丹標準Ⅱ限定至少 3 個親屬罹患結直腸癌或與LS相關的惡性腫瘤(子宮內膜癌、腎盂癌、小腸癌、輸尿管癌)且滿足如下條件:①3名親屬中的1名與其他2名的關系為一級親屬;②至少影響連續 2 代;③至少其中 1名親屬在50 歲前被診斷為結直腸癌;④除外家族性腺瘤性息肉;⑤為病理學確診腫瘤。修訂的 Bethesda 指南診斷依據:①診斷為結直腸癌時小于50 歲;②同時或不同時發生的結直腸癌和其他 LS 相關的癌癥,不限年齡;③診斷為結腸癌時小于60歲且組織學提示MSI 高表達;④結直腸癌患者,其一級親屬有1名或多名患 LS 相關的癌癥,且至少1名親屬發病時小于50歲;⑤結直腸癌患者,其一級或二級親屬中 ≥2名患 LS 相關的惡性腫瘤,年齡不限。滿足以上其中一條的患者建議篩查 IHC或 MSI。如若有證據表明腫瘤組織缺失MMR蛋白(無BRAF基因突變和MLH1基因啟動子甲基化),個人史或家族史達到阿姆斯特丹標準Ⅱ和修訂的 Bethesda 指南的患者,應考慮 LS的可能性,建議進行 LS 相關的基因檢測。LS 相關的卵巢癌有如下特點:①發病年齡相對較小;②漿液性組織類型比例偏高;③發現時很少發生轉移;④生存年限高度依賴分期[7]。
本例患者發病年齡< 50歲,罹患卵巢癌且部分乙狀結腸及直腸有癌浸潤,一級親屬罹患結腸癌時年齡< 50歲,免疫組化證實癌灶中表達 MLH1、PSM2、MSH2蛋白,符合修訂的 Bethesda標準,故建議患者及其家族進行相關疾病的遺傳咨詢及治療。實際上,無論是阿姆斯特丹標準Ⅱ還是修訂的Bethesda指南均會漏診部分LS患者,目前初步篩查LS的方法有MSI檢測、BRAF基因突變檢測、免疫組化[8]。公認最準確可靠的診斷方法為MMR基因檢測,但MMR基因檢測昂貴,目標人群不明確時,難以開展廣泛應用,用免疫組化的方法檢測腫瘤組織中 MMR 蛋白的缺失是一種簡單有效的篩查手段,可作為MSI檢測的替代,若某一蛋白表達缺失,則可選擇進一步檢測確診。目前,國內對于LS相關性卵巢癌研究較少,其發生發展機制及如何進行化學性預防有待進一步研究。
參 考 文 獻
[1] 郭超,劉愛軍. 林奇綜合征相關性子宮內膜癌病理學研究進展. 解放軍醫學院學報,2015,36(6):640-643.
[2] Schmeler KM, Lu KH. Gynecologic cancers associated with Lynch syndrome/HNPCC. Clin Transl Oncol,2008,10(6):313-317.
[3] Sari A, Pollett A, Eiriksson LR, Lumsden-Johanson B, Van de Laar E, Kazerouni H, Salehi A, Sur M, Lytwyn A, Ferguson SE. Interobserver agreement for mismatch repair protein immunohistochemistry in endometrial and nonserous, nonmucinous ovarian carcinomas. Am J Surg Pathol,2019,43(5):591-600.
[4] Ryan NAJ, Evans DG, Green K, Crosbie EJ. Pathological features and clinical behavior of Lynch syndrome-associated ovarian cancer. Gynecol Oncol,2017,144(3):491-495.
[5] Duraturo F, Liccardo R, De Rosa M, Izzo P. Genetics, diagnosis and treatment of Lynch syndrome: old lessons and current challenges. Oncol Lett,2019,17(3):3048-3054.
[6] Syngal S, Brand RE, Church JM, Giardiello FM, Hampel HL, Burt RW; American College of Gastroenterology. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancersyndromes. Am J Gastroenterol,2015,110(2):223-262; quiz 263.
[7] Woolderink JM, De Bock GH, de Hullu JA, Hollema H, Zweemer RP, Slangen BFM, Gaarenstroom KN, van Beurden M, van Doorn HC, Sijmons RH, Vasen HFA, Mourits MJE. Characteristics of Lynch syndrome associated ovarian cancer. Gynecol Oncol,2018,150(2):324-330.
[8] M?ller P, Sepp?l? TT, Bernstein I, Holinski-Feder E, Sala P, Gareth Evans D, Lindblom A, Macrae F, Blanco I, Sijmons RH, Jeffries J, Vasen HFA, Burn J, Nakken S, Hovig E, R?dland EA, Tharmaratnam K, de Vos Tot Nederveen Cappel WH, Hill J, Wijnen JT, Jenkins MA, Green K, Lalloo F, Sunde L, Mints M, Bertario L, Pineda M, Navarro M, Morak M, Renkonen-Sinisalo L, Valentin MD, Frayling IM, Plazzer JP, Pylvanainen K, Genuardi M, Mecklin JP, Moeslein G, Sampson JR, Capella G; Mallorca Group. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut,2018,67(7):1306-1316.
(收稿日期:2019-01-05)
(本文編輯:林燕薇)
