液相色譜-四極桿/靜電場軌道阱質譜鑒定賽拉嗪在人尿中的代謝產物
2019-06-10 07:16:34崔晶晶孫英英沈保華劉萬卉
質譜學報 2019年3期
崔晶晶,孫英英,沈保華,劉萬卉,向 平
(1.煙臺大學藥學院,山東 煙臺 264005;2.司法鑒定科學研究院,上海市法醫學重點實驗室,上海市司法鑒定專業技術服務平臺,上海 200063;3.山東省威海市公安局刑事科學技術研究所,山東 威海 264200)
賽拉嗪(xylazine, C12H16N2S)是一種α2-腎上腺素受體激動劑,它通過刺激中樞神經α2-腎上腺素受體,減少中樞神經系統中去甲腎上腺素和多巴胺的釋放,導致鎮靜、肌肉松弛和對疼痛刺激的感知減少[1],常作為動物的鎮靜劑、止痛藥和肌肉松弛劑,又因其能產生麻醉效果,常被濫用導致中毒事故或犯罪的發生[2]。
藥物輔助犯罪(drug-facilitated crime, DFC)是指在中樞神經抑制劑、興奮劑和致幻劑等精神活性物質影響下,實施的麻醉搶劫、性犯罪等不法行為。隨著網絡傳播和網購的盛行,藥物輔助性犯罪(drug-facilitated sexual assault, DFSA)案件的數量及涉案藥物種類呈上升趨勢[7-8]。DFSA案件因所涉藥物攝入劑量小,代謝快等是法醫毒物鑒定的難點,而尿液中代謝產物檢測時限長的優勢在DFSA案件中發揮了重要作用[7]。賽拉嗪是近兩年常出現于DFSA案件中的藥物之一,但其在人體內的代謝途徑以及主要代謝物的研究資料很少。
目前,代謝物確證研究多采用液相色譜-質譜法[9-10]、氣相色譜-質譜法[11-12]、高效液相色譜-高分辨質譜法[13-14]等。液相色譜-四極桿-靜電場軌道阱質譜(LC-Q-Orbitrap MS)是基于Orbitrap靜電場軌道阱技術測定原理的液質聯用技術,具有高分辨率、高精密度、高靈敏度和動態范圍寬等特點,可同時獲得高質量的多級質譜圖(MSn),保證分析結果的可靠性和準確性[15]。Compound Discoverver軟件包含豐富的工具集,可以進行預期化合物搜索及使用碎片離子搜索闡明(FIsh)結構,將多個樣品數據信息合并到一份報告中,可對研究藥物的體內代謝物提供技術支持。
本研究擬采用LC-Q-Orbitrap MS法及Compound Discoverver軟件對賽拉嗪陽性尿液進行篩查,并對其代謝物進行結構解析,探索賽拉嗪在人體內的代謝方式及途徑。
1 實驗部分
1.1 主要儀器與裝置
UltiMate 3000 UHPLC 液相系統,ThermoScientificTMQ-ExactiveTM組合型四極桿Orbitrap質譜儀(LC-Q-Orbitrap MS):美國Thermo Fisher Scientific公司產品,配有電噴霧離子源(HESI-Ⅱ)及XcaliburTM3.1工作站;TDZ4-WS離心機:上海盧湘儀離心機有限公司產品;XW-80A渦旋混合器:上海醫大儀器有限公司產品;Milli-Q超純水機:美國Millipore公司產品;Minspin高速離心機:德國Eppendorf公司產品;超聲波清洗器:深圳市潔盟清洗設備有限公司產品。
1.2 主要材料與試劑
甲醇、乙腈:色譜純,美國Fisher Scientific公司產品;乙酸乙酯:分析純,上海凌峰化學試劑有限公司產品;甲酸、乙酸銨:瑞士Fluka公司產品;實驗用水:超純水,經Mili-Q系統制備;其他試劑均為分析純。
陽性尿液來自一例DFSA案件,受害人尿液經GC/MS分析檢出賽拉嗪成分。
1.3 樣品前處理
1.3.1液-液萃取法 取1.0 mL待測尿液于5 mL具塞試管中,加入2.0 mL乙酸乙酯提取溶劑,渦旋振蕩2 min后,以離心半徑 12 cm,3 000 r/min離心3 min。轉移有機層,取上層有機溶液,加入一滴NaOH(10%)溶液,重復上述步驟。合并上層有機溶液,于40 ℃空氣流下吹干,向殘留物中加入100 μL甲醇復溶后,待進樣分析。
1.3.2蛋白沉淀法 取100 μL待測尿樣于1.5 mL離心管中,加入900 μL乙腈,渦旋振蕩2 min后離心3 min,上清液轉移至進樣襯管,取5 μL進樣分析。
1.4 實驗條件
1.4.1色譜條件 Thermo Scientific Hypersil GOLD PFP色譜柱(100 mm×2.1 mm×3 μm),流動相:A為5 mmol/L乙酸銨-0.1%甲酸水溶液,B為乙腈;梯度洗脫程序:0~0.5 min(98%A),0.5~10 min(98%~2%A),10~15 min(2%A),15~20 min(98%A);進樣量5.0 μL;流速400 μL/min;柱溫26.7 ℃。
1.4.2質譜條件 電噴霧電離正離子模式(ESI+);噴霧電壓3.5 kV;殼氣、輔助加熱氣、碰撞氣:均使用高純氮氣;探頭加熱器溫度300 ℃;毛細管溫度325 ℃;鞘氣流速35 arb,輔助氣流速10 arb。采用一級全掃描和自動觸發二級(full scan-ddMS2)的數據采集模式。
一級掃描分辨率70 000,二級掃描分辨率17 500,全質量掃描范圍m/z70~1 050,可視寬度為0.7,碰撞能量為40、60、80 eV。進樣前用校正液對質譜的質量軸進行校正,質量數偏差小于5×10-6。
利用XcaliburTM3.1工作站及Compound DiscovererTM3.0軟件進行數據處理。
2 結果與討論
2.1 液-液萃取法
陽性尿液經液-液萃取法前處理后,由LC-Q-Orbitrap MS分析,其全掃描總離子流圖(TIC)示于圖1。賽拉嗪及各代謝產物的高分辨質譜數據列于表1。賽拉嗪的保留時間為3.96 min,準分子離子峰[M+H]+為m/z221.110 58,其MS2譜圖中的碎片離子為m/z164.052 76、147.091 69、90.037 21。m/z90.037 21可作為噻嗪環的特征碎片離子。對于大多數代謝物,因其分子質量太小而不能進一步碎裂,得不到MS3譜圖,所以主要利用代謝物的MS和MS2譜圖進行結構解析。賽拉嗪及各代謝產物(M1~M5)的MS2譜圖示于圖2。通過Compound Discoverer 軟件分析得到5個可疑分子離子峰。準分子離子峰[M+H]+m/z237.105 48,保留時間為3.37 min,比原準分子離子大16 u,可能是增加了1個O為代謝產物M1。M1的[M+H]+離子在MS2譜圖中對應的主要碎片離子為m/z163.086 55、180.047 76,比母離子的MS2碎片離子(m/z147.091 69、164.052 76)大16 u,而m/z90.037 21沒有發生改變,即噻嗪環沒有開裂,進一步說明羥基化反應發生在2,6-二甲基苯胺環上,但難以確認羥基化的具體位點,故推測存在羥基化的兩個同分異構體(M1-1、M1-2)。
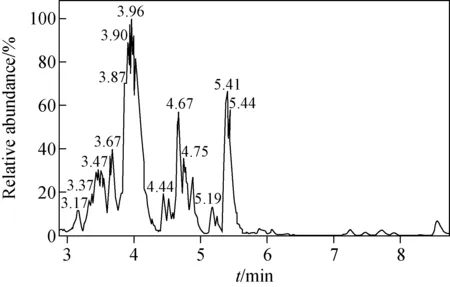
圖1 樣品經液-液萃取處理后的總離子流色譜圖Fig.1 Total ion current chromatogram of the sampleprepared by liquid-liquid extraction
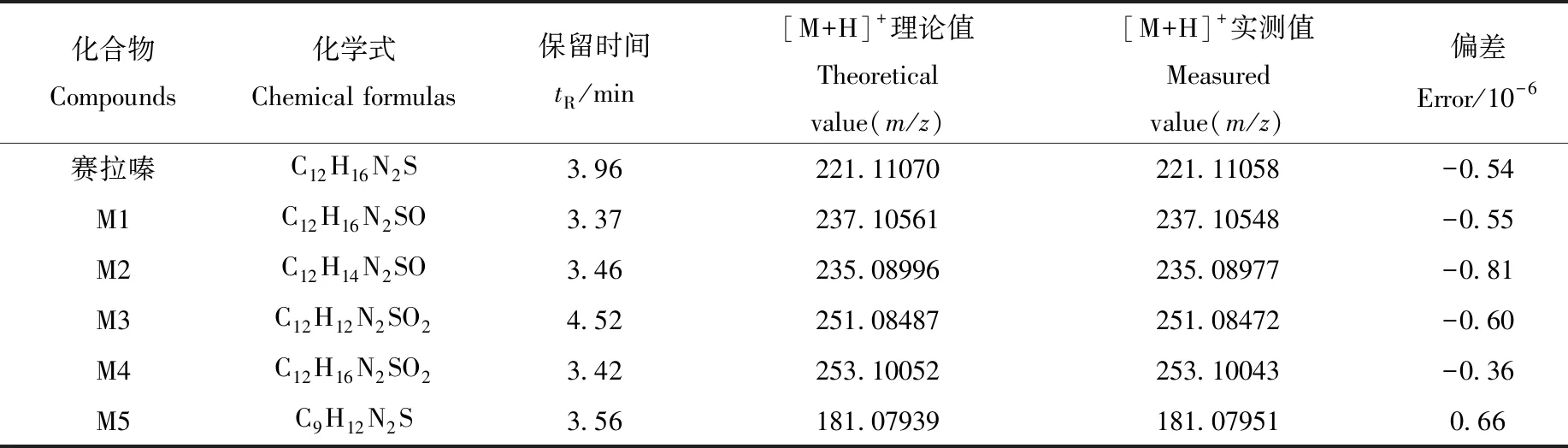
表1 賽拉嗪在人尿液中代謝產物的LC-Q-Orbitrap MS數據(液-液萃取方法)Table 1 LC-Q-Orbitrap MS data of xylazine and its metabolites in human urine(liquid-liquid extraction)
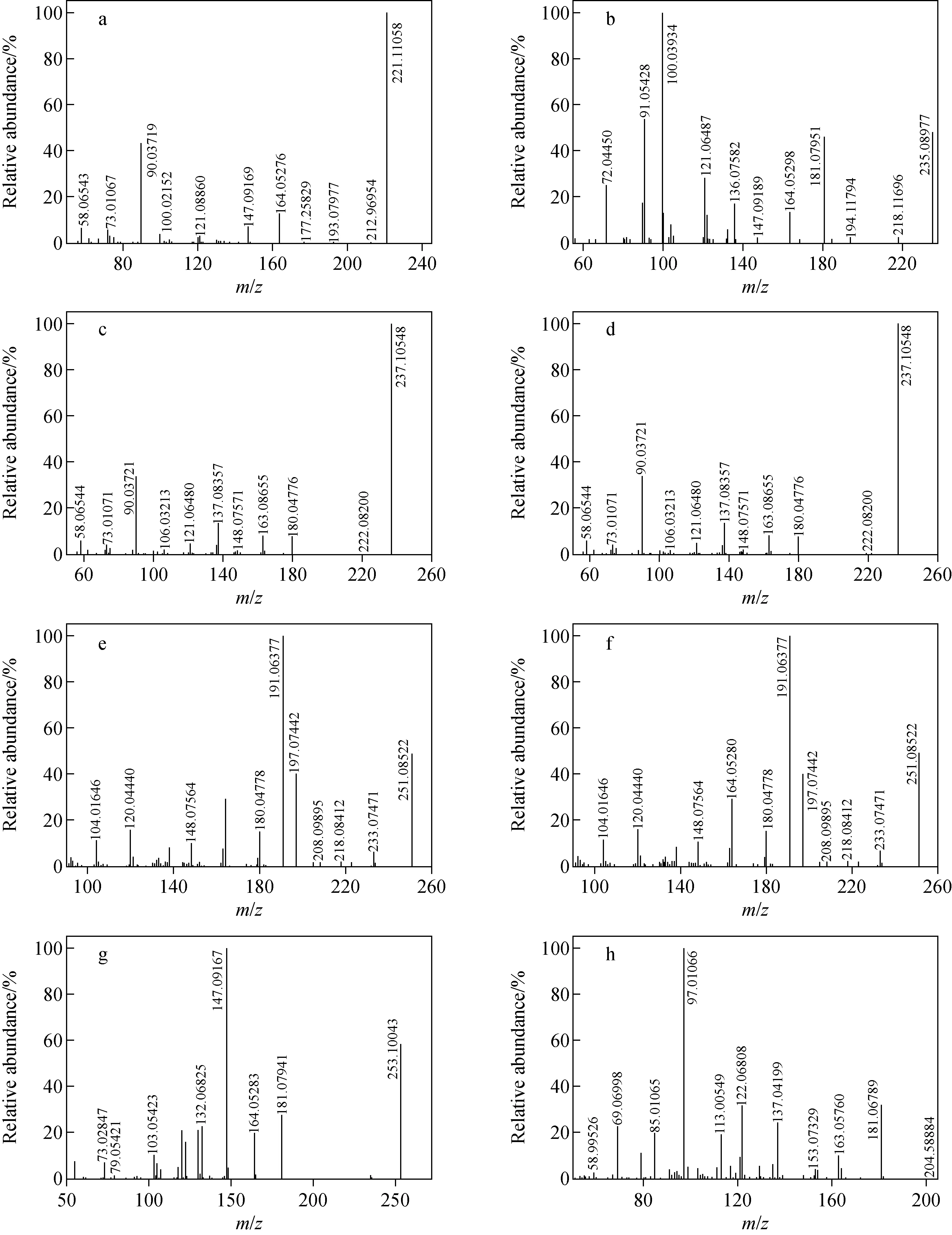
注:a.賽拉嗪;b.M2;c.M1-1;d.M1-2;e.M3-1;f.M3-2;g.M4;h.M5圖2 賽拉嗪及各代謝產物MS2譜圖(液液萃取法)Fig.2 MS2 spectra of xylazine and its metabolites by liquid-liquid extraction
準分子離子峰[M+H]+m/z235.089 77,保留時間為 3.46 min,比原準分子離子大14 u(+16-2),推測為賽拉嗪的氧化代謝產物M2。M2的[M+H]+離子在MS2譜圖中對應的主要碎片離子為m/z181.079 51、164.052 98、136.075 82,未觀察到在m/z90.037 21 處噻嗪環的特征碎片離子,表明噻嗪環發生了氧化開環。
準分子離子峰[M+H]+m/z251.084 72,保留時間為4.52 min,比原準分子離子大30 u(16+16-2),推測為賽拉嗪的氧化羥基化代謝產物M3。M3的[M+H]+離子在MS2譜圖中的主要碎片離子m/z104.016 44比對應原體的碎片離子m/z90.037 21大了14 u(+16-2),表明噻嗪環發生了氧化反應。MS2譜圖中m/z197.074 25、148.075 64比M2的碎片離子m/z181.079 51、132.080 7大了16 u,表明在2,6-二甲基苯胺環處發生了羥基化反應。由于難以確認發生羥基化的具體位點,故推測存在氧代羥基化的兩個同分異構體(M3-1、M3-2)。
準分子離子峰[M+H]+m/z253.100 43,保留時間為3.42 min,比原準分子離子大32 u(16+16),表明存在發生二羥基化或S-氧化成砜反應的代謝產物M4。MS2譜圖存在m/z147.091 67、164.052 83處的豐富碎片離子及2,6-二甲基苯胺環的特征碎片離子m/z181.079 41,排除2,6-二甲基苯胺環的羥基化反應,推斷該反應發生在噻嗪環上。因未發生水的消除反應,排除了噻嗪環的二羥基化反應,因此推測M4為賽拉嗪S-氧化成砜的代謝產物。
準分子離子峰[M+H]+m/z181.079 91,保留時間為3.56 min,比原準分子離子小40 u(—C3H4),推測為賽拉嗪經N,S-脫烷基化生成N-(2,6-二甲基苯基)硫脲,為代謝產物M5。在MS2譜圖中,可以觀察到m/z122.096 46處的豐富碎片離子,應是賽拉嗪N-脫烷基化生成的代謝產物2,6-二甲基苯胺,m/z181.079 91可作為2,6-二甲基苯胺的前體離子存在,因其分子質量較小,未能直接檢測出來。
2.2 蛋白沉淀法
由蛋白沉淀法前處理后的陽性尿液經LC-Q-Orbitrap MS進樣后,出現多個色譜峰,其TIC色譜圖示于圖3。賽拉嗪的保留時間是4.10 min,準分子離子峰m/z221.110 60,其MS2譜圖中主要特征碎片離子為m/z164.052 76、147.091 67、90.037 19,示于圖4。通過Compound Discoverer 軟件分析,得到7個分子離子峰。賽拉嗪及各代謝產物對應的高分辨質譜數據列于表2,其中準分子離子m/z237.105 45、253.100 27的主要碎片離子與液-液萃取法得到的代謝產物M1、M4一致,推斷其結構相同。準分子離子峰m/z165.102 28,保留時間為3.07 min,與賽拉嗪相差56 u(+O—C3H4S),經Compound Discoverer軟件分析為代謝產物M6,主要碎片離子為m/z148.075 82、122.096 44,推測其是由N-(2,6-二甲基苯基)硫脲分解得到的尿素結構。
準分子離子峰m/z413.135 74,保留時間為3.12 min,比原體增加了192 u,為代謝產物M7,與M1的準分子離子相比相差176 u(+C6H8O7),推測M7為M1與葡糖醛酸共軛得到的Ⅱ相代謝產物。
準分子離子峰m/z317.061 04,保留時間為3.64 min,比賽拉嗪原體增加了96 u(+SO3),為代謝產物M8,其主要碎片離子為m/z90.037 20,表明噻嗪環沒有改變,m/z237.105 47與M1的準分子離子峰相同,故推斷M8(M8-1/M8-2)是由M1(M1-1/M1-2)與硫酸共軛得到的Ⅱ相代謝產物。
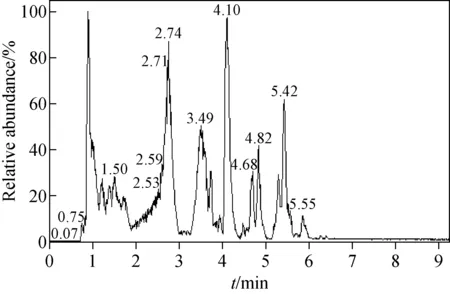
圖3 經蛋白沉淀后的總離子流色譜圖Fig.3 Total ion current chromatogram of the sampleprepared by protein precipitation
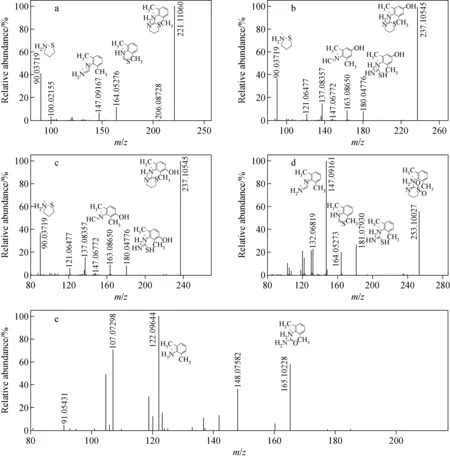
a.賽拉嗪;b.M1-1;c.M1-2;d.M4;e.M6圖4 賽拉嗪及代謝產物經蛋白沉淀后得到的MS2譜圖Fig.4 MS2 spectra of xylazine and its metabolites by protein precipitation
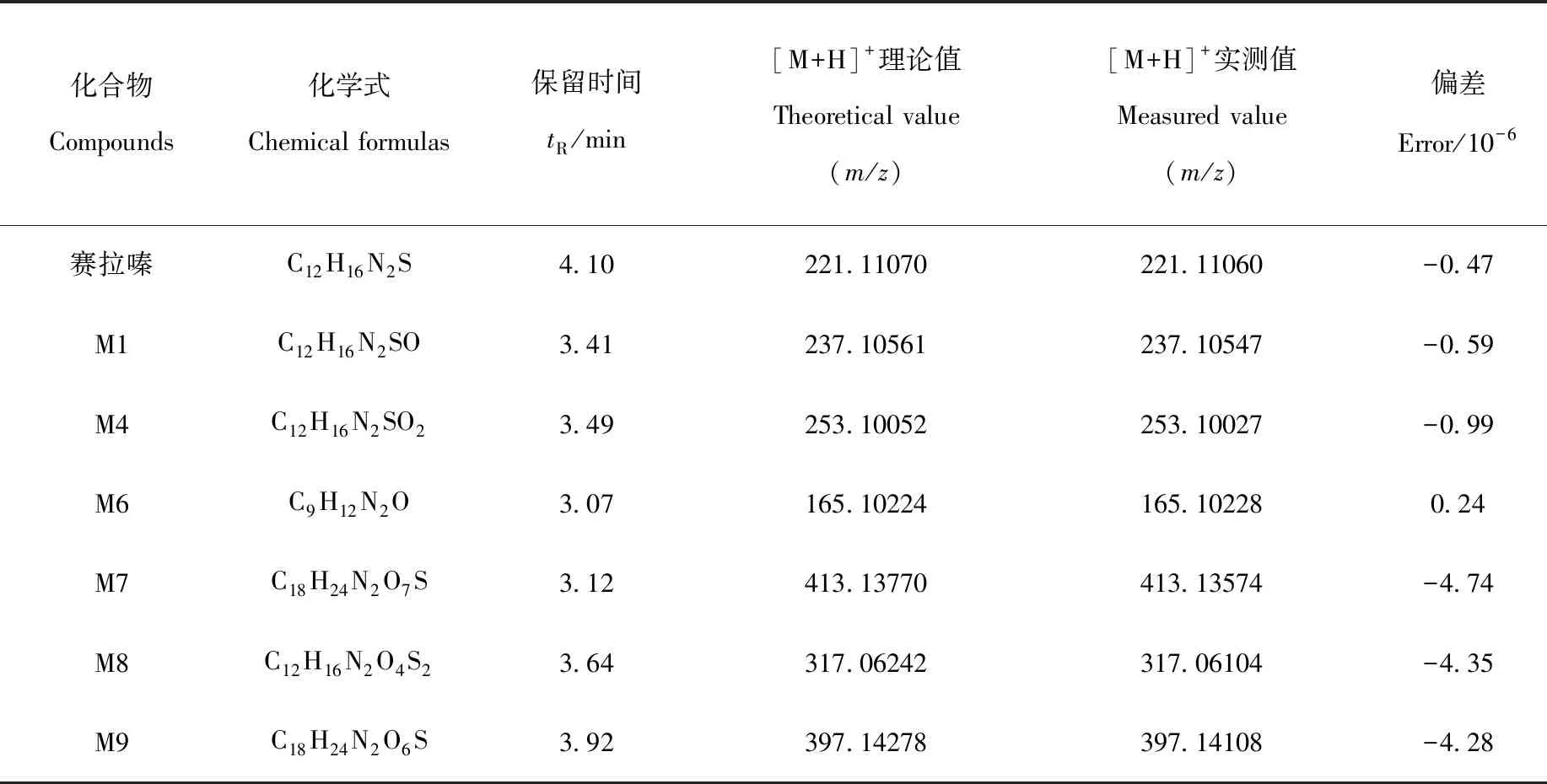
化合物Compounds化學式Chemical formulas保留時間tR/min[M+H]+理論值Theoretical value(m/z)[M+H]+實測值Measured value(m/z)偏差Error/10-6賽拉嗪C12H16N2S4.10221.11070221.11060-0.47M1C12H16N2SO3.41237.10561237.10547-0.59M4C12H16N2SO23.49253.10052253.10027-0.99M6C9H12N2O3.07165.10224165.102280.24M7C18H24N2O7S3.12413.13770413.13574-4.74M8C12H16N2O4S23.64317.06242317.06104-4.35M9C18H24N2O6S3.92397.14278397.14108-4.28
準分子離子峰m/z397.141 08,保留時間為3.92 min,比賽拉嗪原體增加了176 u(+C6H8O6),為代謝產物M9,推測為賽拉嗪發生N-葡糖醛酸結合反應得到的Ⅱ相代謝產物。
2.3 討論
本實驗利用液-液萃取法鑒定到賽拉嗪的7個代謝產物,M1-1、M1-2為羥基化產物,M2為氧化產物,M3-1、M3-2為氧代羥基化產物,M4為S-氧化產物,M5為N-(2,6-二甲基苯基)硫脲。經M5的MS2譜圖推導所得的代謝產物2,6-二甲基苯胺與相關文獻報道一致[9-10]。利用蛋白沉淀法鑒定到賽拉嗪的8個代謝產物,M1-1、M1-2為羥基化產物,M4、M6為S-氧化產物,M7、M8-1、M8-2、M9均為Ⅱ相代謝產物。蛋白沉淀的前處理方法能夠得到更多強極性的Ⅱ相代謝產物,而液-液萃取法更容易得到水溶性強的Ⅰ相代謝產物。兩種方法相互補充,能夠得到更充分的數據闡明藥物的體內代謝途徑。
根據鑒定所得的代謝產物結構可以推斷賽拉嗪及其代謝產物之間的生物轉化途徑,示于圖5。轉化途徑與大鼠尿液[13]、大鼠肝微粒體[14]、馬尿[12]等的研究結果相似,主要的代謝物包括羥基化產物、氧化產物及S-氧化產物等。Lavoie等[14]在大鼠肝微粒體中檢測到的N-羥基化產物在人尿液中沒有檢測到,可能由物種不同或者濃度較低造成的。Meyer等[13]用標準尿液篩查方法(standard urine screening protocol)鑒定一例高加索人陽性案例的結果與本實驗結果一致,可以認為不同人群賽拉嗪的體內代謝途徑一致。經研究,建議在生物檢材賽拉嗪分析時增加主要代謝物M1為目標物,以增強可靠性,延長檢測時限,提供更豐富的藥動學數據。含有硫脲結構的藥物具有一定的毒性,賽拉嗪的代謝產物N-(2,6-二甲基苯基)硫脲的毒理學研究對于中毒后的監測具有重要的臨床意義。
3 結論
本研究利用液-液萃取法和蛋白沉淀法對賽拉嗪陽性尿液進行前處理,通過LC-Q-Orbitrap MS進行解析,共鑒定到13種代謝產物。賽拉嗪在人體內發生羥基化、氧化、N-脫烷基化、S-氧化等Ⅰ相代謝反應,羥基化代謝產物可與葡萄糖醛酸及硫酸結合發生Ⅱ相代謝反應。本研究初步闡明了賽拉嗪在人體內的代謝途徑和主要代謝產物。
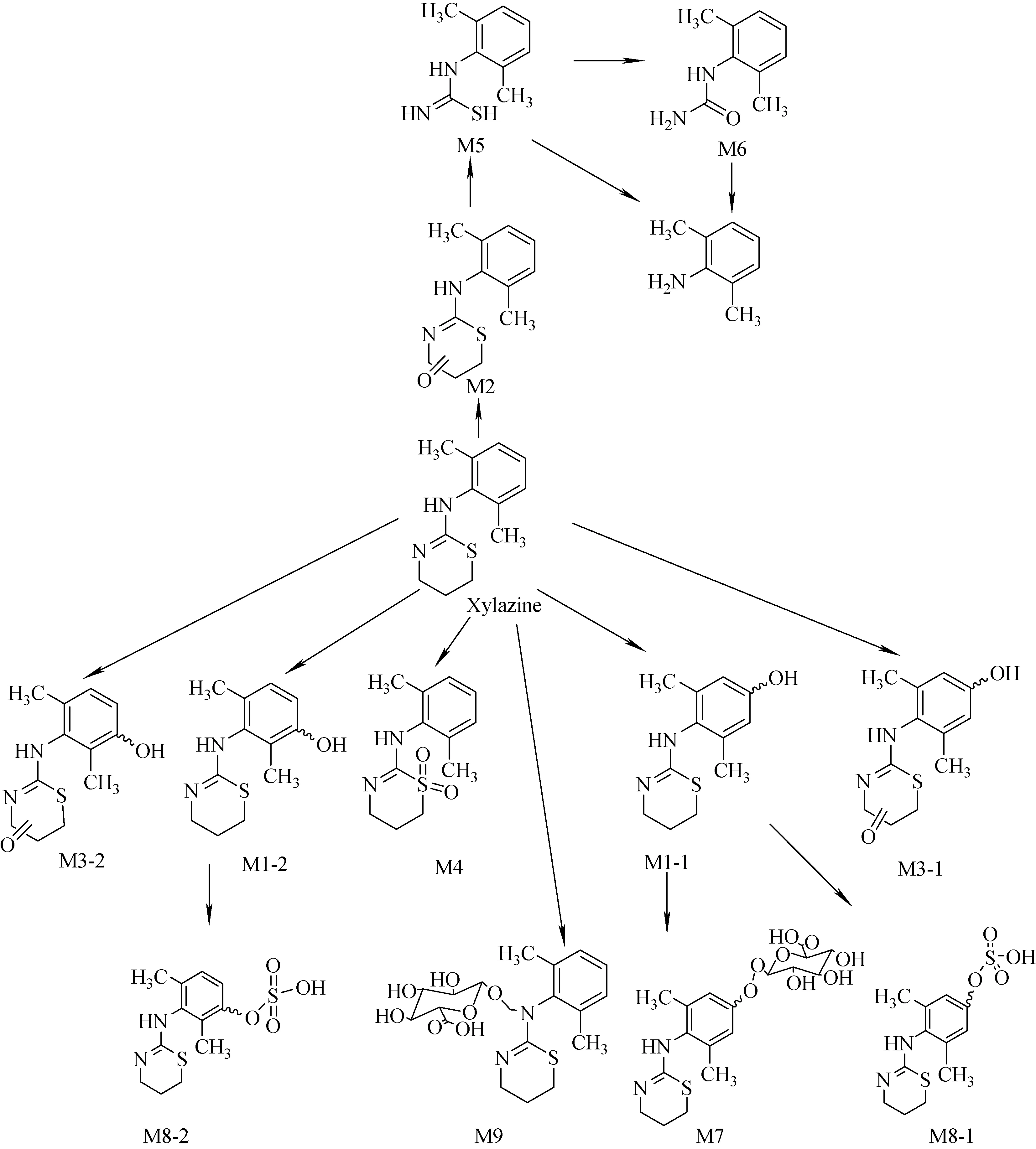
圖5 賽拉嗪在人體內的代謝途徑Fig.5 Proposed metabolic pathways of xylazine in humans
