Sin-QuEChERS結合UPLC-MS/MS同時檢測茶葉中10種有機磷農藥殘留
2019-06-10 07:16:34郝東宇劉松南席興軍潘燦平張維冰
質譜學報 2019年3期
蘭 韜,初 僑,郝東宇,,劉松南,席興軍,潘燦平,張維冰
(1.中國標準化研究院,北京 100191;2.齊齊哈爾大學,黑龍江 齊齊哈爾 161006;3.北京市茶葉質量監督檢驗站,北京 100162;4.中國農業大學,北京 100083)
茶葉是我國傳統飲品,富含茶多酚、氨基酸、黃酮、茶多糖等營養物質[1],具有降低膽固醇、提高免疫力、減肥瘦身等保健功效,深受人們的喜愛。但茶葉中農藥殘留問題制約了我國茶葉產業的發展[2-5]。有機磷農藥是一類含有磷原子的有機酯類化合物或硫羥衍生物,因其具有藥效高、品種多、防治范圍廣、成本低、選擇性好、藥害小、可降解、殘毒低等優點,已成為茶葉種植中廣泛使用的一類農藥[6-7]。目前,各類有機磷農藥中毒事件頻發,如日本的馬拉硫磷污染食品事件[8]、印度的久效磷中毒慘案[9]等,都說明了有機磷農藥的殘留毒害問題日益嚴重,需要加大對其管控的力度。
近年來,我國對茶葉中有機磷農藥殘留進行了較嚴格的規定。如2017年實施的GB 2763—2016[10]中規定:甲胺磷等農藥的限量值為0.05 mg/kg,接近大部分檢測方法的極限。由于有機磷農藥具有升溫分解的特性,這對農殘提取凈化方法的準確度、靈敏度提出了更高的要求。茶葉中含有大量的茶多酚、咖啡因、葉綠素等多酚、嘌呤和色素類物質[11],具有很強的基質效應,嚴重影響茶葉中痕量農藥殘留的檢測[12]。目前,茶葉中農藥殘留檢測的樣品前處理方法主要有固相萃取法[13]、固相微萃取法[14]、液-液萃取法[15]、加速溶劑萃取法[16]、基質固相分散法[17]等,這些傳統方法在一定程度上能夠滿足基本的茶葉樣品前處理需求,但操作方法以及試劑消耗還有待簡化。
QuEChERS(Quick、Easy、Cheap、Effective、Safe)方法是由美國農業部Anastassiades教授于2003年開發的一種用于農產品檢測的快速樣品前處理技術[18]。即先將樣品粉碎,然后用乙腈提取,加入無水MgSO4等鹽類去除水分,再加入乙二胺-N-丙基硅烷(PSA)等吸附劑除雜后,取上清液進行檢測。該方法具有簡便、回收率高等優點[19],結合高靈敏的液相色譜-質譜聯用法,已成為目前農藥和獸藥殘留檢測的主流方法[20-29]。
Sin-QuEChERS方法是對QuEChERS方法的改進,即采用Sin-QuEChERS凈化小柱可對樣品溶液實現一步凈化[30],結構圖示于圖1。將具有較大比表面積的多壁碳納米管與PSA、C18等固相材料及優化柱體結構相結合,制成Sin-QuEChERS小柱。將樣品置于離心管后,加入乙腈渦旋振蕩、離心,將Sin-QuEChERS小柱插入離心管內,按壓Sin-QuEChERS小柱得上清凈化液,凈化液可直接進樣。該方法能夠改善基質干擾的去除效果,如對色素、脂類、部分糖類甾醇類等的凈化效果有明顯提高,色譜圖干擾明顯減小。與其他提取凈化方法相比,具有操作簡單、對雜質凈化效果明顯、對農藥吸附少、結果準確等優點,能夠極大地縮短分析時間。同時,由于該方法不涉及加熱環節,非常適用于分析升溫分解的有機磷農藥。目前,Sin-QuEChERS前處理技術還處于起步階段,尚未被用于藥物殘留(特別是有機磷農藥)檢測領域。
本研究擬采用Sin-QuEChERS方法結合超高效液相色譜-串聯質譜(UPLC-MS/MS)法,檢測茶葉中有機磷農藥殘留。針對茶葉樣品具有較強基質效應以及有機磷農藥升溫易分解的特點,對Sin-QuEChERS樣品前處理技術進行改進。選取GB 2763—2016中對茶葉制定了限量要求的氧化樂果(OME)、敵百蟲(DIP)、甲胺磷(MET)、內吸磷(DEM)等4種有機磷農藥,以及樂果(DIM)、亞胺硫磷(PHO)、馬拉硫磷(MAL)、喹硫磷(QUI)、敵敵畏(DIC)、久效磷(MON)等6種容易在茶樹上超范圍使用的有機磷農藥為研究對象,進行UPLC-MS/MS檢測。希望為快速、簡易、準確地評價茶葉的質量安全奠定基礎。
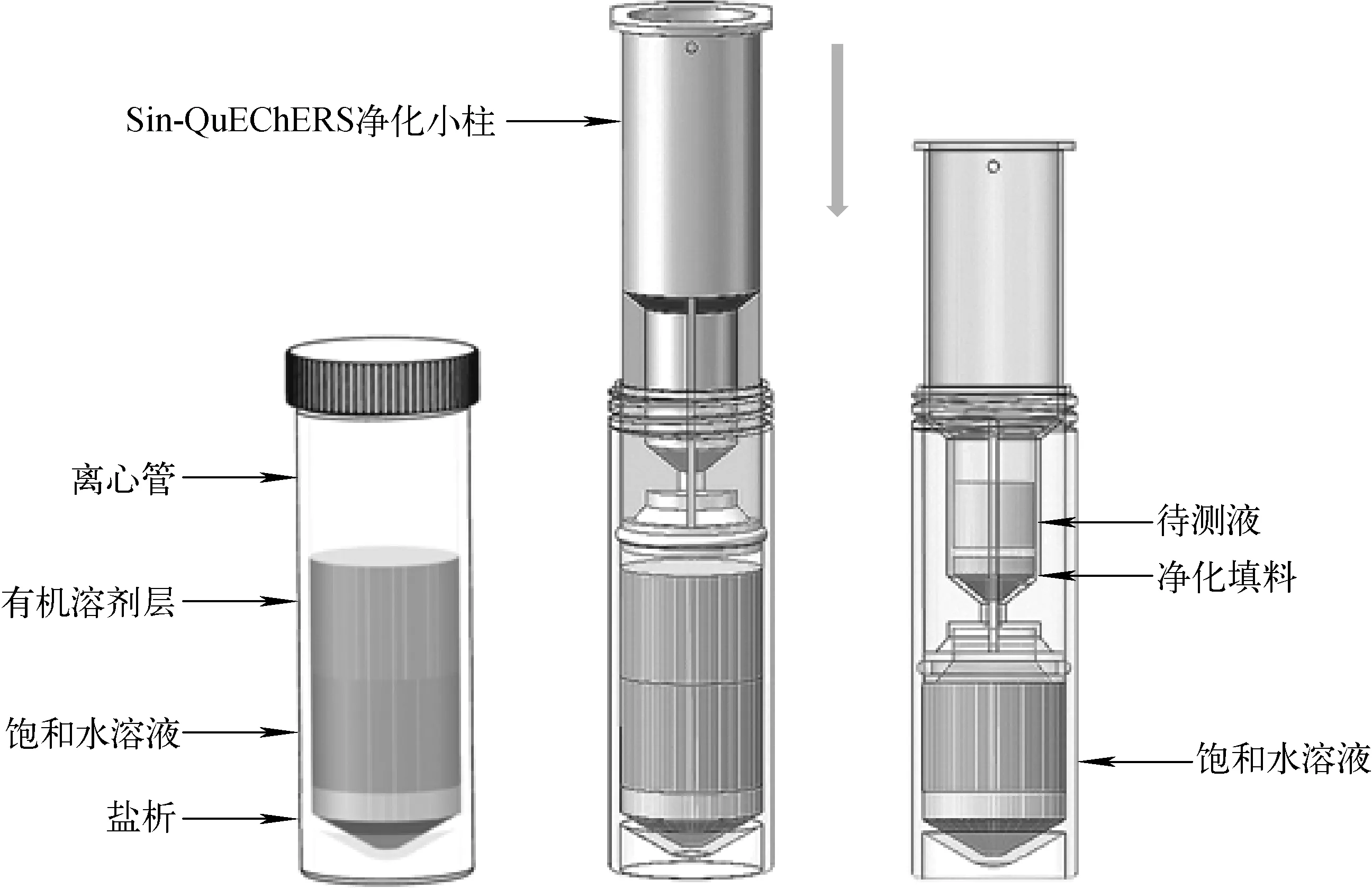
圖1 Sin-QuEChERS凈化小柱結構圖Fig.1 Diagram of the Sin-QuEChERS purification column
1 實驗部分
1.1 主要儀器與裝置
ACCELA高效液相色譜儀、TSQ Quantum Access MAX三重四極桿質譜儀:賽默飛世爾科技有限公司產品;高速萬能粉碎機:天津市泰斯特儀器有限公司產品;循環水式多用真空泵:鄭州長城科工貿有限公司產品;旋轉蒸發器:上海亞榮生化儀器廠產品;固相萃取儀:上海汗諾儀器有限公司產品;VM200旋渦振蕩儀:托摩根生物科技有限公司產品;JJ 500電子天平(感量0.01 g):常熟雙杰測試儀器廠產品;Eppendorf Research移液器(量程20~200 μL):Eppendorf中國有限公司產品;Sin-QuEChERS固相萃取小柱:北京綠綿科技有限公司產品。
1.2 主要材料與試劑
空白茶葉樣品(不含待檢10種有機磷農藥):婺源茗眉綠茶樣品,由江西省出入境檢驗檢疫局技術中心提供;樂果、氧化樂果、亞胺硫磷、馬拉硫磷、喹硫磷、敵百蟲、甲胺磷、內吸磷、敵敵畏和久效磷標準品:濃度均為100 mg/L,北京壇墨質檢科技有限公司產品,于4 ℃冰箱保存;乙腈和甲醇:色譜純,德國Merck公司產品;甲酸(分析純):上海阿拉丁生化科技股份有限公司產品。
1.3 茶葉中有機磷農藥殘留的前處理方法
分別稱取20 g各品種茶葉,經粉碎后過80目篩,裝袋密封備用,于4 ℃冷藏保存。
提取方法:稱取3 g(精確至0.01 g)粉碎后的茶葉粉末于50 mL離心管中,加入10 mL乙腈,旋渦振蕩2 min,以5 000 r/min離心5 min。
凈化方法:將Sin-QuEChERS小柱插入離心后的離心管內,按壓Sin-QuEChERS小柱直至小柱內的待測液達4 mL,取小柱內的上清液過0.22 μm有機濾膜后上機檢測。
1.4 溶液配制
空白基質溶液:稱取3 g(精確至0.01 g)空白茶葉樣品,采用Sin-QuEChERS方法對此樣品進行粉碎、提取、凈化,所得上清液即為空白基質溶液,將該溶液置于4 ℃冰箱保存,備用。
農藥標準儲備溶液:分別取100 μL樂果、氧化樂果、亞胺硫磷、馬拉硫磷、喹硫磷、敵百蟲、甲胺磷、內吸磷、敵敵畏和久效磷的標準品于10 mL容量瓶中,用乙腈定容至刻度線,配制1 000 μg/L的10種有機磷農藥混合標準溶液,現配現用。
基質農藥標準儲備溶液:分別取100 μL樂果、氧化樂果、亞胺硫磷、馬拉硫磷、喹硫磷、敵百蟲、甲胺磷、內吸磷、敵敵畏和久效磷等標準品于10 mL容量瓶中,以空白基質溶液定容至刻度線,配制1 000 μg/L的10種有機磷農藥混合標準品,現配現用。
1.5 標準曲線
用乙腈將標準儲備溶液稀釋成1、5、10、50、100、250、500 μg/L,經HPLC-MS/MS分析,繪制標準儲備溶液的標準曲線。
用空白基質溶液將基質標準儲備溶液稀釋成1、5、10、50、100、250、500 μg/L,經HPLC-MS/MS分析,繪制基質標準儲備溶液的標準曲線。
1.6 加標回收實驗
稱取3 g(精確至0.01 g)空白茶葉樣品粉末,分別加入60、300、900 μL農藥標準儲備溶液,使粉末中農藥含量分別達到20、100、300 μg/kg,按照1.3節方法,將上清液過0.22 μm有機濾膜后上機檢測。
1.7 色譜條件
Zorbax SB-C18色譜柱(4.6 mm×250 mm×5 μm);柱溫25 ℃;進樣量10 μL;流速1.8 mL/min;流動相:A為0.1%甲酸-水溶液,B為乙腈;梯度洗脫程序:0~6 min(5%~40%B),6~9 min(40%~80%B),9~10 min(80%~100%B),10~13 min(100%B)。
1.8 質譜條件
電噴霧離子源正離子模式,多反應監測(MRM)模式掃描,霧化氣為液氮,毛細管溫度320 ℃,霧化氣溫度300 ℃,鞘氣壓力6 125 kPa,輔助氣壓力875 kPa,離子噴霧電壓3 200 V,CID壓力0.2 kPa。
2 結果與討論
2.1 色譜條件的優化
本研究選用三通管路分流模式,當柱流速為1.8 mL/min時,流入質譜端的流速為0.39 mL/min,既滿足了質譜儀對流速的要求,又能在保證柱壓處于安全范圍內的前提下,達到快速分離的目的。所以本實驗選用流速1.8 mL/min。
本研究對比了乙腈-0.1%甲酸水溶液和甲醇-0.1%甲酸水溶液兩種流動相體系對10種有機磷農藥的分離效果,結果示于圖2。在乙腈-0.1%甲酸水溶液流動相體系下,各組分出峰時間較快,峰形尖銳,10種農藥組分均達到基線分離,示于圖2a。而在甲醇-0.1%甲酸水溶液流動相體系下,各組分出峰時間延遲,峰展寬明顯,分離度較差,示于圖2b。綜上,本研究選用乙腈-0.1%甲酸水溶液流動相體系進行后續實驗。
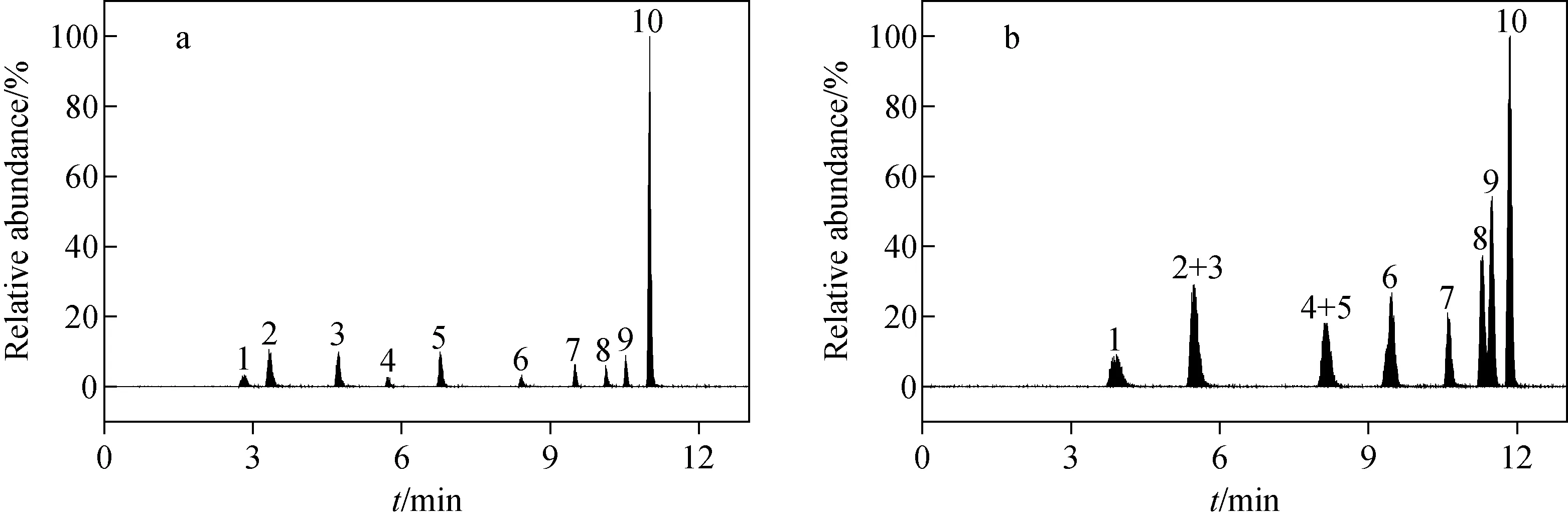
注:a.乙腈-0.1%甲酸水溶液;b.甲醇-0.1%甲酸水溶液;1.甲胺磷;2.氧化樂果;3.久效磷;4.敵百蟲;5.樂果;6.敵敵畏;7.內吸磷;8.亞胺硫磷;9.馬拉硫磷;10.喹硫磷圖2 兩種流動相體系對10種有機磷農藥分離對比Fig.2 Separation and comparison of 10 kinds of organophosphorus pesticides by two mobile phase systems
本研究所涉及的其他色譜條件,如色譜柱填料、色譜柱溫等均參考國家標準GB 23200.13—2016[31],其中流動相梯度洗脫程序在此標準基礎上進行了微調,以滿足各種農藥的基線分離,具體條件參見1.7節。
2.2 質譜條件的優化
本實驗向流動相中加入0.1%甲酸以提高目標物的離子化程度。校準儀器后,取農藥標準品,用乙腈稀釋成1 mg/L的標準溶液,經蠕動泵注入質譜儀中,選擇MRM模式,并選取各農藥的2對離子對,系統自動得到優化的碰撞能和套管透鏡補償電壓,10種有機磷農藥的質譜參數列于表1。
2.3 提取方法的優化
目前,在茶葉農殘檢測中,對提取方法中是否使用水存在較多爭議。有研究者[32]認為,當樣品含水量少于25%時,需要補充水分來增加細胞通透性,提高前處理過程中農藥殘留的析出。但向樣品中加水易造成大量基質溶出,影響檢測的準確性。國家標準GB 23200.13—2016《食品安全國家標準茶葉中448種農藥及相關化學品殘留量的測定 液相色譜-質譜法》[31]和AOAC發布的食品中農藥殘留檢測方法[33],均直接通過乙腈提取樣品。另外,龐國芳等[34]通過比對16個實驗室的檢測結果,證實茶葉中農藥殘留提取無需加水。為驗證提取樣品過程是否需要加水,本研究基于Sin-QuEChERS方法,分別考察了樣品加水和不加水的提取效果。選取加標水平均為300 μg/L的10種有機磷混合標準品,第一組為不加水,以10 mL乙腈提取;第二組為加水提取,當加入5 mL水時,不足以浸潤3 g茶葉樣品,當加入10 mL水時,會稀釋樣品,所以,選擇加入7 mL水和10 mL乙腈作為提取溶劑。兩組的后續凈化方法一致,回收率列于表2。
由表2可見,是否加水對方法回收率的影響顯著,不加水與加水提取的回收率分別為71.6%~99.6%、47.5%~116.9%,且不加水提取3次測試結果的相對標準偏差(RSD)明顯優于加水提取。
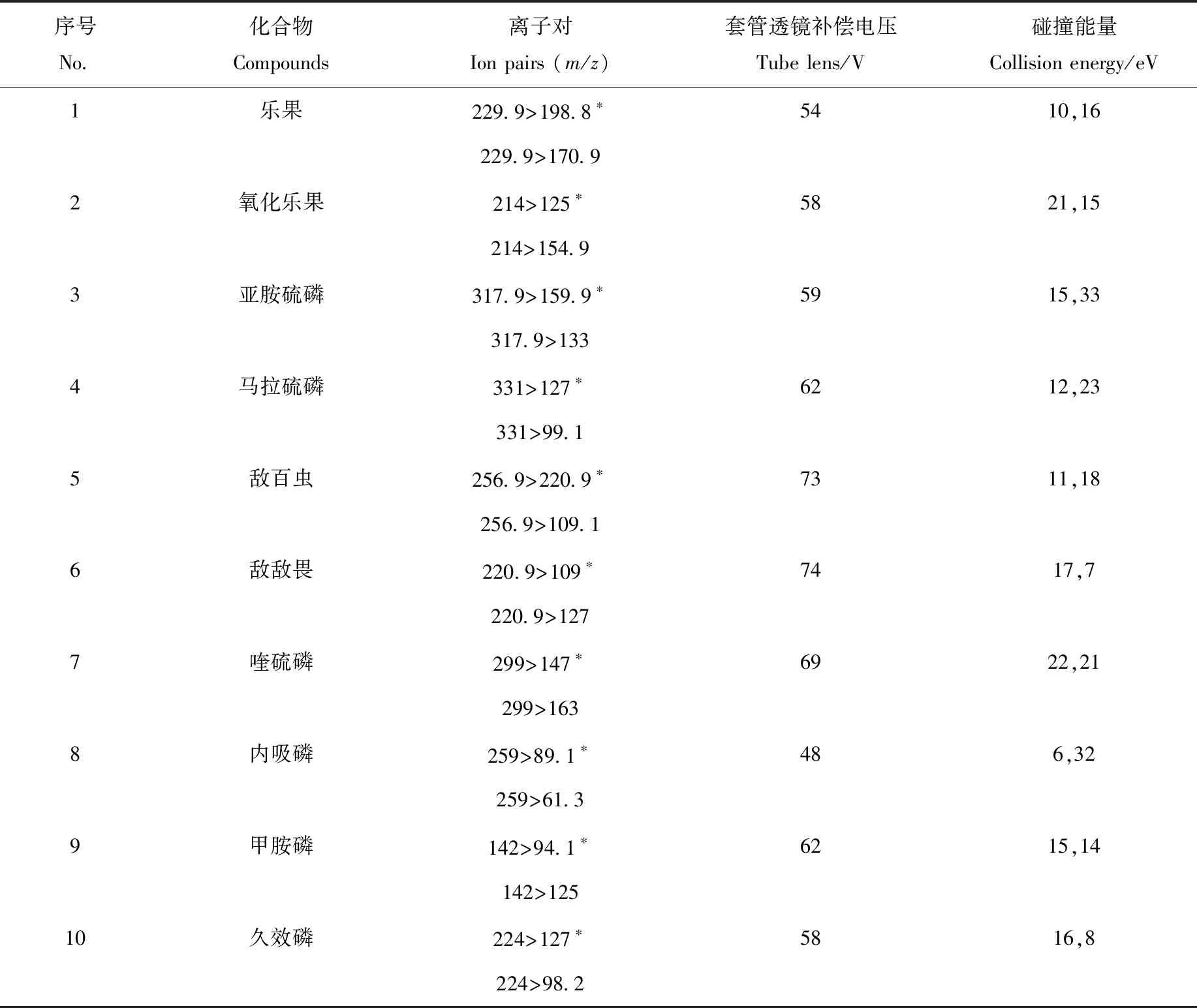
表1 10種有機磷農藥的質譜參數Table 1 Mass spectrometric parameters of 10 organophosphorus pesticides
注:*表示定量離子對
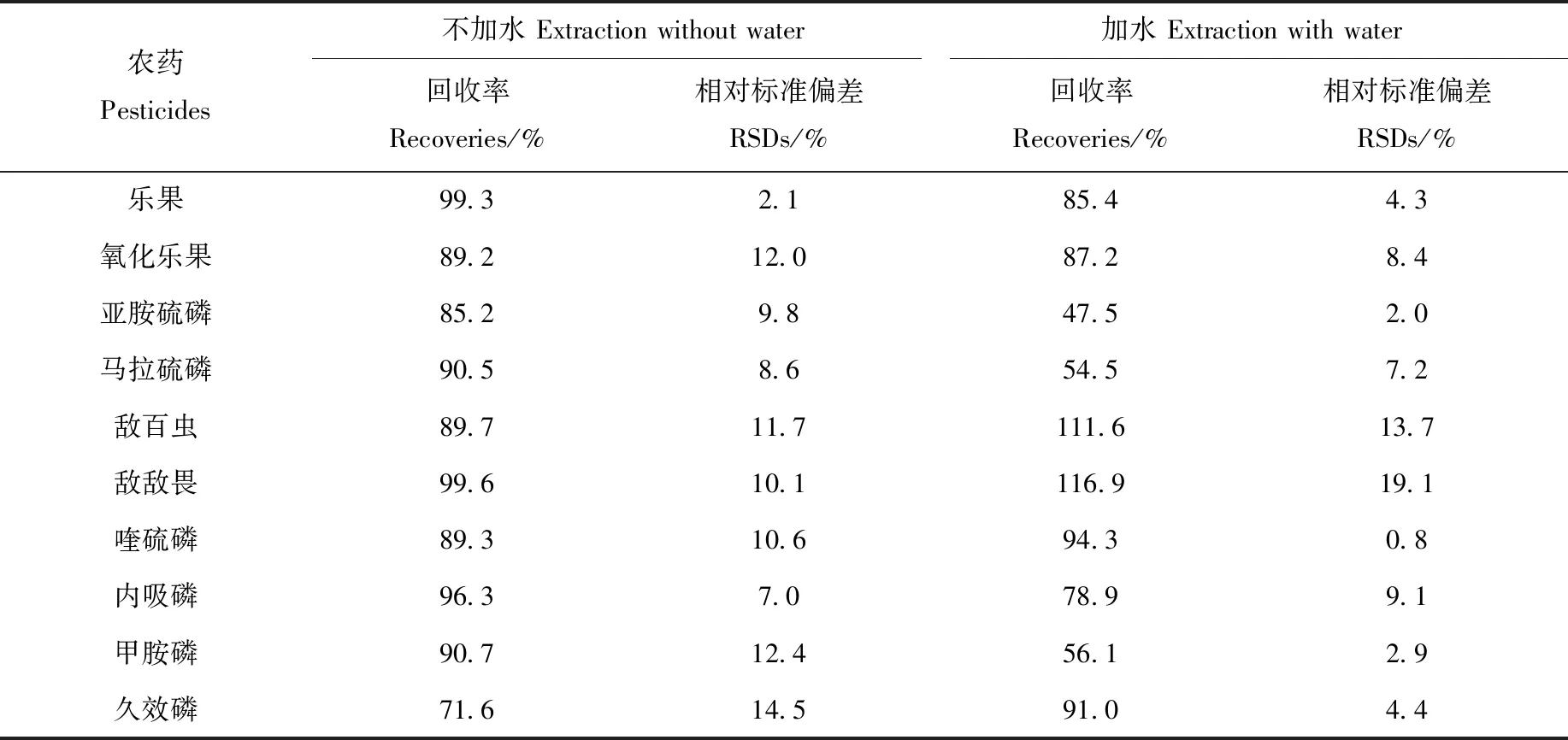
表2 加水和不加水提取的回收率對比Table 2 Comparison of recoveries of Sin-QuEChERS method with water extraction and without water extraction
以滇紅茶一等品為例,按照Sin-QuEChERS的提取方法對茶葉進行提取,比較加水與不加水提取時的溶液顏色。結果表明,加水提取的上層清液顏色明顯比不加水提取的顏色深,說明加水提取后大量色素類物質溶出,其基質影響明顯高于不加水提取,不利于后續的凈化過程,造成提取回收率較差,所以本實驗采用不加水提取的方法。
2.4 基質效應
在實際農藥殘留檢測中,當存在基質效應時,使用純溶劑標準曲線進行校準,常會引起分析結果偏差和樣品回收率的錯誤計算。對基質效應不明顯的化合物,基質標準曲線與溶劑標準曲線基本重合。本研究通過對比不含基質的標準工作溶液標準曲線(A)和含基質(濃度3 g/mL)的標準工作溶液標準曲線(B)的斜率,以表征10種有機磷農藥的基質效應,結果列于表3。結果表明,敵敵畏、喹硫磷、馬拉硫磷、內吸磷、樂果等5種農藥的基質效應不明顯,敵百蟲、甲胺磷、亞胺硫磷等3種農藥存在明顯的基質減弱效應,氧化樂果存在一定的基質增強效應,久效磷的基質增強效應非常明顯。鑒于部分農藥存在較明顯的基質效應,后續均需基于基質溶液進行實驗。
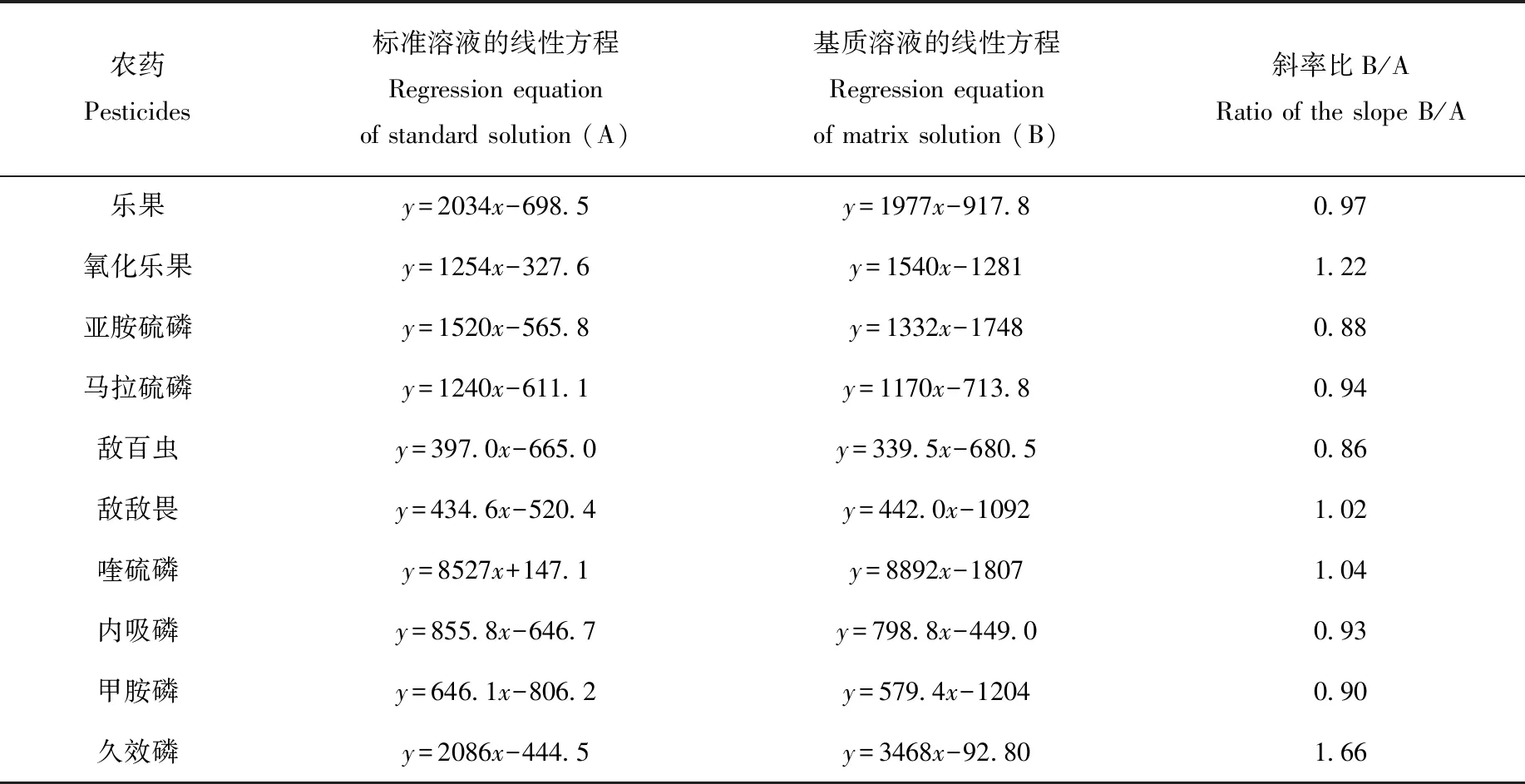
表3 10種有機磷農藥的基質效應對比Table 3 Comparison of matrix effect on 10 organophosphorus pesticides
2.5 方法的線性范圍、靈敏度、檢出限、定量限
10種有機磷農藥的線性方程、線性范圍、檢出限、定量限列于表4。10種有機磷農藥3次檢測的線性范圍均為1~500 μg/L,峰面積與樣品濃度之間的線性關系良好,線性相關系數在0.999 4~1.000 0之間。以定量離子對的3倍信噪比(S/N)確定方法的檢出限(LOD)為0.08~1.38 μg/kg,以定量離子對的10 倍信噪比(S/N)確定方法的定量限(LOQ)為0.27~4.60 μg/kg。
2.6 方法的回收率與精密度
為驗證方法的可靠性,本研究對空白茶葉樣品進行加標回收實驗,加標水平分別為20、100和300 μg/kg的混合標準品,每個加標水平做5次平行實驗,方法回收率和精密度列于表5。加標水平為20、100、300 μg/kg時,10種農藥的平均回收率為77.6%~110.7%、74.6%~108.4%、71.6%~99.6%,RSD分別小于13.6%、11.9%、14.5%。該結果表明,本研究建立的Sin-QuEChERS方法在不同加標水平下,平均回收率均處于71.6%~115.0%之間,RSD小于14.5%,具有較高的回收率和良好的精密度,能夠滿足多種茶葉農殘檢測的要求。
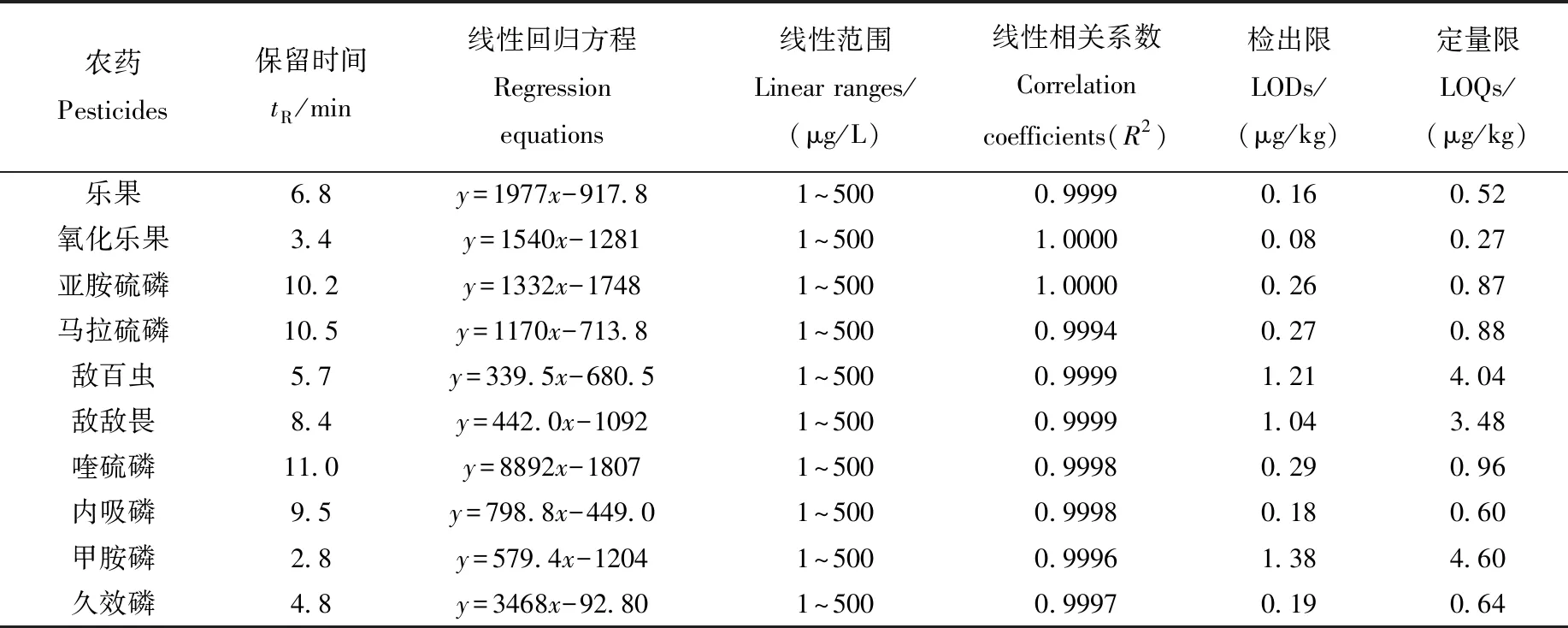
表4 10種有機磷農藥的線性方程和線性范圍(n=3)Table 4 Linear equations, linear ranges for 10 organophosphorus pesticides (n=3)
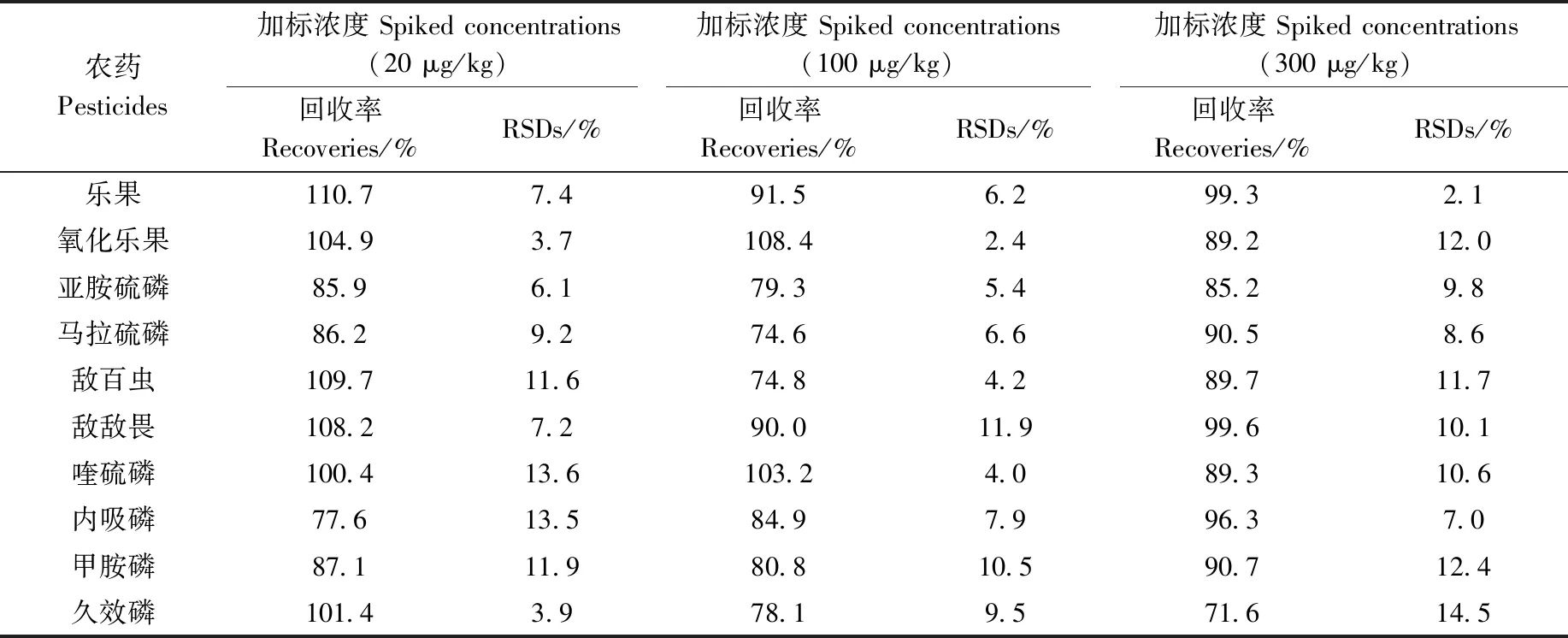
表5 方法的回收率(n=5)Table 5 Recoveries of the method for adding pesticides (n=5)
2.7 Sin-QuEChERS方法與國家標準方法的比較
以綠茶為例,將國家標準方法GB 23200.13—2016[31](敵敵畏參照SN/T 1950—2007行業標準[35])中的10種有機磷農藥的回收率作為理論數值,考察Sin-QuEChERS方法的回收率,結果列于表6。結果表明,Sin-QuEChERS方法的回收率在71.6%~115.0%之間,國家標準方法的回收率在60.9%~118.5%之間,二者的回收率差異較小。在國家標準方法中,每個樣品需要增加旋轉蒸發步驟,整個樣品前處理時間一般需要約1.5 h。而在本研究中,Sin-QuEChERS方法的樣品前處理僅需10 min,可在0.5 h內完成1個樣品檢測。該方法既保證了實驗結果的穩定性,又大大提升了檢測效率。另外,Sin-QuEChERS方法提供了久效磷的回收率范圍,可為標準方法提供補充。
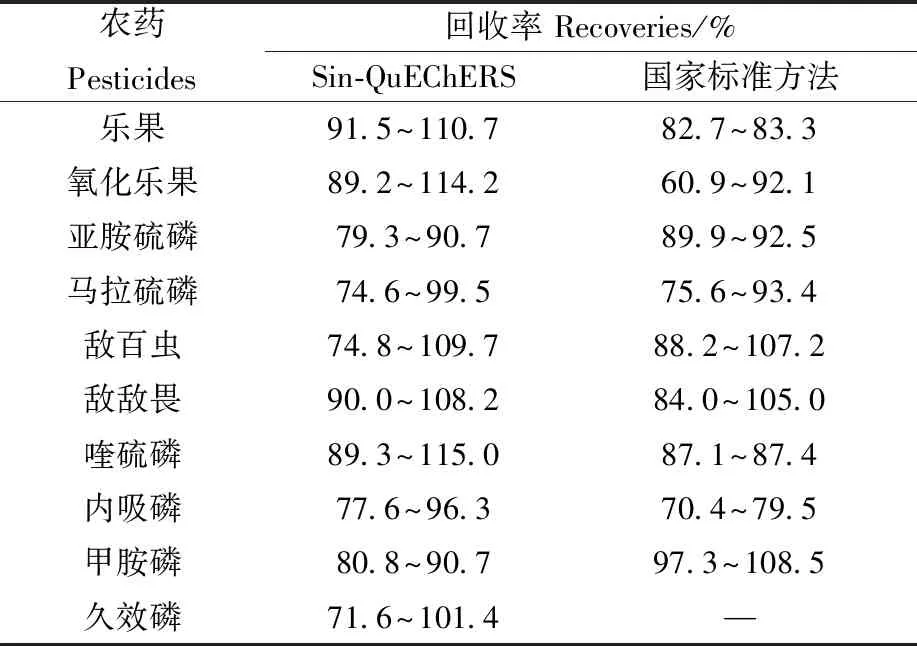
表6 Sin-QuEChERS 方法和國家標準方法對10種有機磷農藥回收率的對比Table 6 Comparison of recoveries of 10 pesticeids between Sin-QuEChERS and the national standards
2.8 實際樣品檢測
采用本研究建立的Sin-QuEChERS結合HPLC-MS/MS方法,檢測市售的黃山毛峰、碧螺春、龍井茶、祁門紅茶、滇紅茶、正山小種紅茶、白毫銀針、白牡丹、安吉白茶、安化黑茶一等品和二等品共20種茶葉中的10種有機磷農藥,結果列于表7。
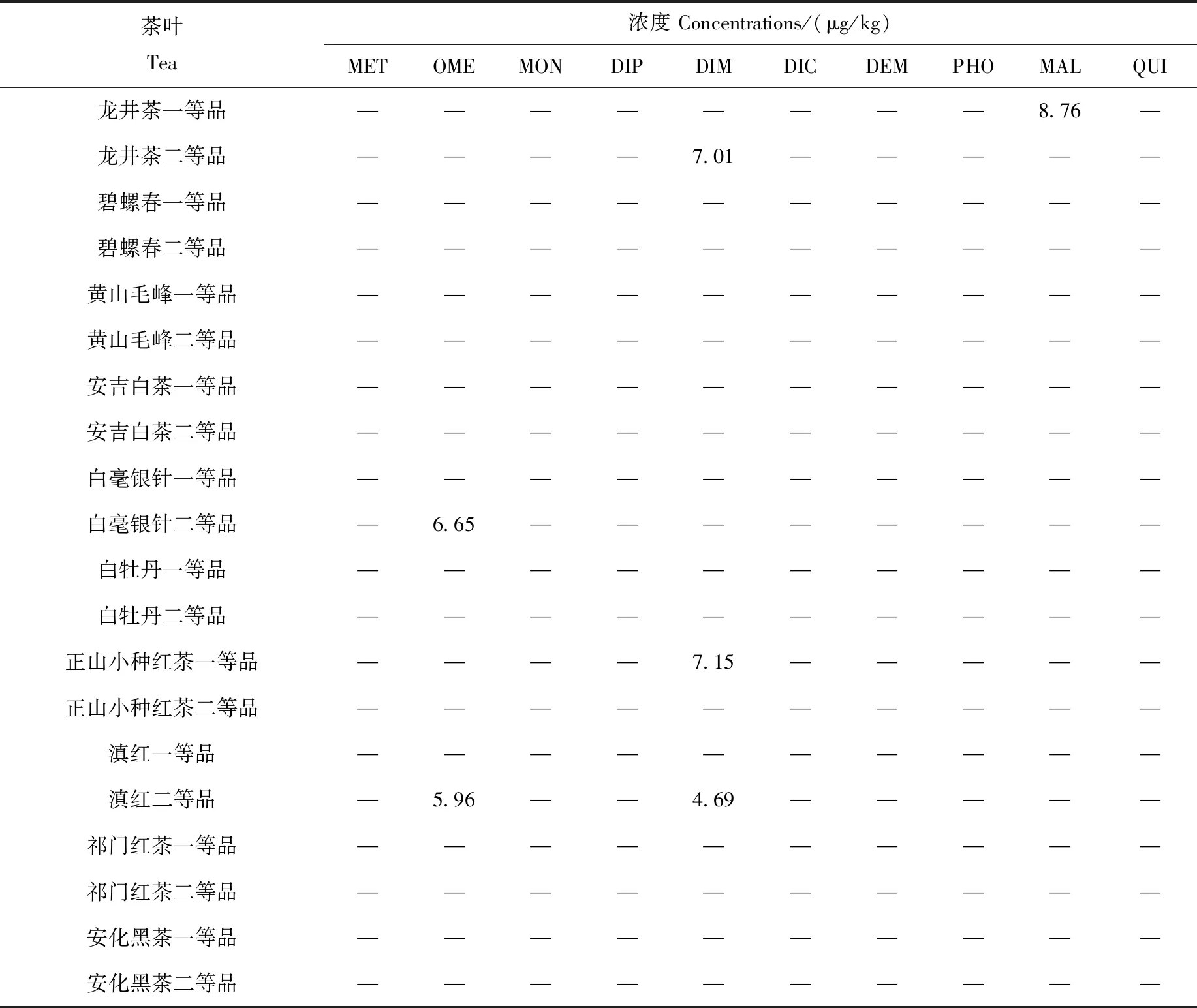
表7 20種茶葉樣品中有機磷農藥的檢測結果Table 7 Detection results of organophosphorus pesticides in 20 kinds of tea
注:—表示未檢出
由表7可見,市售茶葉中有機磷農藥的檢出量較少,均低于歐盟標準中最高0.010 mg/kg的限量要求,說明我國對于茶葉中有機磷農藥的管控較嚴格。
3 結論
本研究針對茶葉樣品具有較強的基質效應以及有機磷農藥升溫易分解的特點,對Sin-QuEChERS樣品前處理技術進行改進,并結合UPLC-MS/MS技術建立了同時檢測茶葉中10種有機磷農藥殘留的方法。在優化的Sin-QuEChERS方法條件和UPLC-MS/MS檢測條件下,樣品前處理時間可控制在10 min以內,每個樣品能夠在0.5 h內完成分析。10種有機磷農藥的線性范圍為1~500 μg/L,檢出限為0.08~1.38 μg/kg,定量限為0.27~4.60 μg/kg。在不同的加標水平下,平均回收率均在71.6%~115.0%之間,RSD小于14.5%,具有較高的回收率和良好的精密度。與傳統樣品預處理方法相比,本方法在保證了方法的靈敏度、精密度和普適性的基礎上,極大地提升了方法的易用性和分析效率。將該方法應用于空白茶葉樣品加標分析和20種市售茶葉中有機磷農藥的檢測,證明了方法的可行性。該方法能夠為快速、簡易和準確地檢測茶葉中多種有機磷農藥殘留提供新途徑。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
意林原創版(2016年10期)2016-11-25 10:28:30
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
專用汽車(2016年4期)2016-03-01 04:13:43
