3β-羥基-Δ5-C27類固醇脫氫酶缺陷1例病例報告
2019-06-04 07:23:22田培超趙彩紅史丹丹石小亞
中國循證兒科雜志 2019年2期
張 碧 田培超 趙彩紅 王 越 史丹丹 石小亞 姚 運
1 病例資料
女,4歲,因“凝血功能異常伴肝脾腫大3年”于2018-1-8入鄭州大學第一附屬醫院(我院)兒內科。患兒1歲7個月時(2014-9-6)因“頭皮血腫反復不愈”至我院就診,凝血功能指標見表1,肝功能未見異常(表2),考慮“凝血因子K缺乏”,予“肌注維生素K1、輸注新鮮冰凍血漿”后頭皮血腫消失,凝血功能改善(表1,2014-9-11)。1年前無誘因鼻衄至當地醫院就診,查PLT 65×109·L-1,骨髓細胞學提示PLT減少,肝腎功能指標均在正常值范圍,遂至我院。
注 正常參考值:凝血酶原時間(PT) 9.6~14 s,PT% 70%~130%,活化部分凝血活酶時間(APTT)23~35 s, 纖維蛋白原(FIB)2~4 g·L-1, 凝血酶時間(TT)14~21 s, 纖維蛋白原降解產物(FDP)0~5 μg·mL-1,D-二聚體 0~0.55 mg·L-1
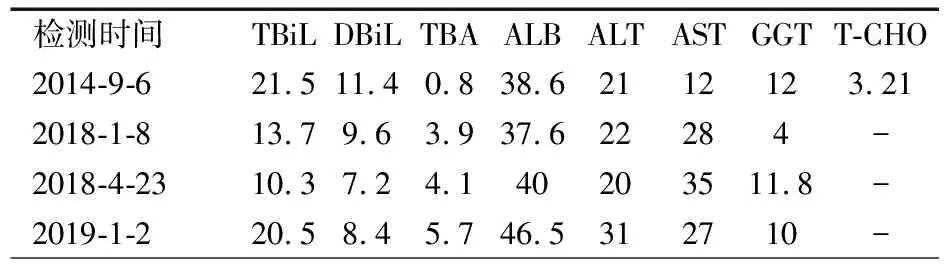
表2 患兒血生化指標檢測結果
注 正常參考值:總膽紅素(TBiL)0~25 μmol·L-1,直接膽紅素(DBiL)0~10 μmol·L-1,總膽汁酸(TBA)0~20 μmol·L-1,白蛋白(ALB)35~55 g·L-1,ALT、AST:0~40 U·L-1,谷氨酰轉肽酶(GGT)0~58 U·L-1,總膽固醇(T~CHO)<5.2 mmol·L-1;-:未檢測
患兒自起病以來,飲食、睡眠、精神可,體重同正常同齡兒。患兒系G1P1,足月順產,孕期未訴特殊事件,無窒息搶救史,出生體重2 800 g;生后混合喂養,按時預防接種。父母非近親結婚,家族中未見同類病患者。
體格檢查:體重21 kg,身高110 cm。神志清,精神可,營養、發育良好,皮膚黏膜無黃染、皮疹及出血點。心肺檢查未見異常,全腹平軟,無肝掌、蜘蛛痣及腹壁靜脈曲張,肝臟右鎖骨中線肋下捫及4 cm,劍突下捫及4 cm,質地韌,邊鈍,無觸痛。脾臟于左鎖骨中線肋下捫及6 cm,質軟,未觸及異常包塊。神經系統查體未見異常。
輔助檢查:血常規WBC 6.13×109·L-1,Hb 113 g·L-1,PLT 86×109·L-1。凝血酶原時間(PT)22.2 s,PT% 37.0%,活化部分凝血活酶時間(APTT)50.7 s;肝功能(表2)、腎功能、血糖、血脂均正常;腹部彩超示肝臟肋下4 cm,包膜尚光滑,實質回聲增粗;膽壁增厚,內透聲差;脾臟厚徑35 mm,長徑130 mm,肋下65 mm, 實質回聲均勻,脾靜脈增寬;雙腎盞多發強回聲。骨髓細胞檢查:骨髓增生明顯活躍,粒系和紅系比值正常,細胞形態大致正常,色素充盈可;全片可見巨核細胞500個,分類25個,幼稚巨核細胞3個,成熟無PLT形成巨核細胞22個,可見成堆及散在PLT。尿常規、尿有機酸氣相色譜-質譜聯合檢測、血氨基酸和酰基串聯質譜檢測均未見異常膽汁酸代謝產物。
為進一步明確診斷,經家屬知情同意后用EDTA抗凝管采集患兒及其父母外周靜脈血各2 mL,用DNA提取試盒(北京百泰克生物技術有限公司)提取基因組DNA。經二代測序得到的變異序列,通過8WA軟件(版本:0.7.9a)與UCSC hg19參考基因組進行比對,得到致病的突變位點。采用Sanger法驗證突變位點及其父母相對應的基因區域,確定患兒的突變類型和遺傳方式。圖1顯示,患兒檢出HSD3B7基因c.45-46delAG和c.543dupG的復合雜合變異,分別來自父親和母親,符合常染色體隱性遺傳規律。前者為編碼DNA序列上第45~46位置缺失了A和G 2個堿基,導致p.Thr15fs移碼突變;后者為編碼DNA第543位上重復插入1個G堿基,導致p.Leu182fs移碼突變。在人類基因突變數據庫(HGMD)及千人基因組數據庫檢索,c.543dupG目前尚無報道,經MutationTaster在線分析軟件預測該突變為致病性突變;c.45-46delAG已有報道[1]。
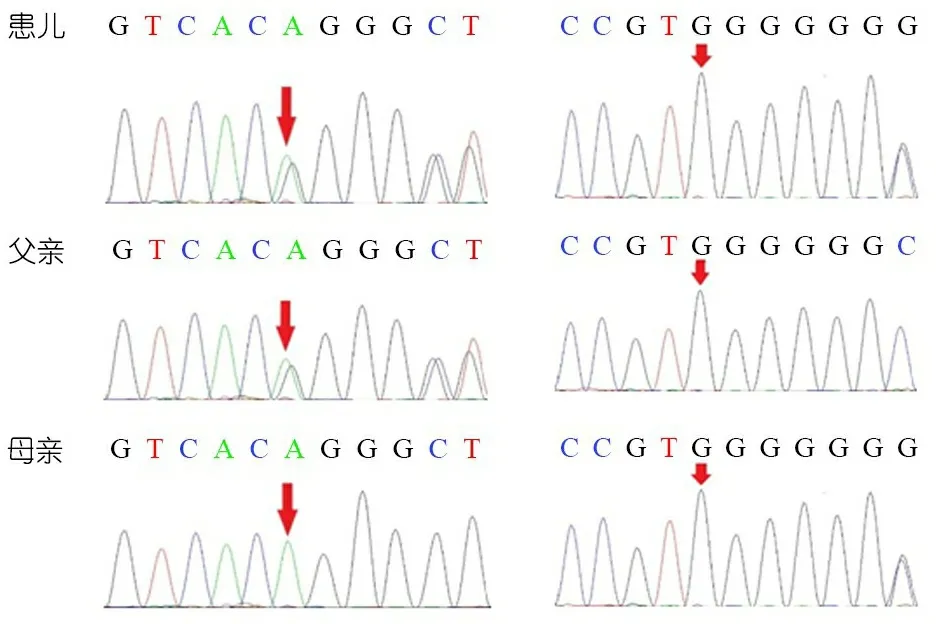
圖1 患兒及其父母HSD3B7基因測序結果
注 患兒為c.45-46delAG和c.543dupG的復合雜合變異,患兒父親攜帶HSD3B7基因的雜合變異c.45-46delAG,患兒母親攜帶1個HSD3B7基因的雜合變異c.543dupG
患兒以PLT減少起病,主要考慮血液系統疾病,未給予特殊治療。基因確診3β-羥基-Δ5-C27類固醇脫氫酶缺陷,予鵝去氧膽酸(CDCA)10 mg·kg-1·d-1,tid;脂溶性維生素A、D、E和K。治療1個月后(表1,2018-4-23)復查凝血功能基本正常。出院后隨訪至2019-1-2,血常規及凝血功能正常(表1);腹部彩超:肝實質光點增粗,肋下約3 cm,形態正常,脾臟肋下5 cm,雙腎皮質強回聲。
2 討論
原發性膽汁酸合成障礙是一種罕見的常染色體隱性遺傳性疾病,由于膽汁酸固醇環的修飾和側鏈的氧化不全所致,占新生兒及兒童不明原因膽汁淤積癥的1%~2%[2, 3]。膽汁酸是在肝臟中由膽固醇合成,涉及17種肝酶[4],其中3β-羥基-Δ5-C27類固醇脫氫酶缺陷是初級膽汁酸合成障礙中最常見的酶缺陷。3β-羥基-Δ5-C27類固醇脫氫酶由位于16號染色體16p11.2的HSD3B7基因編碼,是肝臟內膽汁酸合成過程的關鍵酶之一。
1987年Clayton等[5]首次報道了3β-羥基-Δ5-C27類固醇脫氫酶缺陷。檢索萬方數據知識服務平臺、維普期刊資源整合平臺、中國學術期刊網絡出版總庫和PubMed,國內外報道的3β-羥基-Δ5-C27類固醇脫氫酶缺陷患兒共72例,國內文獻14例[1, 6-8],國外文獻58例[5, 9-18]。男42例,女30例,平均年齡4.0歲(1月至17歲)。臨床表現:黃疸46例,佝僂病 18例,脂肪瀉 13例,凝血功能障礙5例;腹部超聲:肝大46例 (其中合并脾大10例),腎臟結構異常14例(2例腎結石、6例多囊腎、2例腎臟結構不清、2例腎臟呈囊性變、2例雙腎多發囊性變并鈣質沉積);在脂肪瀉患兒中有7例患兒出現腱反射消失[19]。16例患兒有家族史(其中4例兄妹)。
72例3β-羥基-Δ5-C27類固醇脫氫酶缺陷患兒起病的嚴重程度不一,Setchell等[4]證實外源性初級膽汁酸會抑制內源性膽汁酸合成途徑,從而抑制內源性毒性膽汁酸產生。實際上,HSD3B7基因敲除小鼠有脂肪和脂溶性維生素吸收障礙且無肝病[20],與本文報道的病例類似,但具體機制尚不清楚。脂溶性維生素吸收障礙作為本病的臨床特點之一,在臨床中常不被重視。本例患兒表現為脂溶性維生素K缺乏引起的凝血功能障礙。3β-羥基-Δ5-C27類固醇脫氫酶缺陷患兒有神經系統累及,7例患兒出現腱反射消失,且均合并脂肪瀉,考慮與維生素E缺乏有關,具體機制不詳[19]。當體內維生素D缺乏明顯時,部分患兒以行走不穩、佝僂病起病[13, 21]。目前尚無維生素A缺乏相關癥狀報道。
14例腎臟結構異常形成機制尚無明確解釋,予初級膽汁酸替代治療后雙腎結構恢復正常[1, 18],考慮是原發病導致的。也有文獻報道就診時年齡大的患兒,更易出現腎臟結構改變,可能與膽汁酸合成障礙時產生的不飽和膽汁酸的慢性毒性有關[1]。本文病例腎臟彩超顯示雙腎盞多發強回聲,以后的隨訪中會繼續關注腎臟的變化,若隨著患兒凝血功能及肝脾的好轉,腎臟改變也逐漸減輕甚至恢復正常,則說明腎臟結構改變系原發病導致。
目前針對3β-羥基-Δ5-C27類固醇脫氫酶缺陷的治療大多是經驗性的。僅使用CDCA[1, 13]、CDCA+膽酸片(CA)[13]、熊去氧膽酸(UDCA)+CDCA[14-16]、單獨使用CA[12, 15],均被認為有效。Gonzales等對13例該病患者進行了長達20余年的隨訪,發現口服CA是安全有效的長期治療方法[12, 14]。本文病例予CDCA 10 mg·kg-1·d-1,tid及脂溶性維生素A、D、E、K,1月后復查凝血功能基本正常,1年后復查腹部彩超發現肝脾略有縮小,腎臟未見明顯改變。
UDCA作為親水性膽汁酸,可以降低膽汁酸誘導的肝細胞膜損傷的風險,臨床上常首先以UDCA作為治療膽汁淤積癥的經驗性用藥,且發現臨床癥狀和生化指標改善顯著。推測UDCA可能由于利膽作用暫時緩解部分患兒的臨床癥狀,但由于UDCA不能抑制膽固醇7α-羥化酶活性,不能減少異常的膽汁酸生成,不建議長期應用其治療3β-羥基-Δ5-C27類固醇脫氫酶缺陷。初級膽汁酸(CA及CDCA)作為替代治療的原發性膽汁酸,可以進入肝腸循環,并通過負反饋抑制膽固醇7α-羥化酶活性,激活負反饋調節膽汁酸合成,抑制肝毒性代謝產物的產生,提供足夠大的膽汁酸池刺激膽汁流動,最后增加管腔內濃度,使脂肪和脂溶性維生素正常化吸收。初級膽汁酸替代療法最重要的作用是恢復對膽汁酸合成的正常生理反饋。
3β-羥基-Δ5-C27類固醇脫氫酶缺陷作為可以被治療的罕見的遺傳性疾病,臨床表現較為多樣,在嬰兒期常以黃疸為首發癥狀,同時血清中GGT及總膽汁酸正常或減低,伴有肝脾腫大、脂肪瀉、生長發育遲緩、佝僂病、凝血功能障礙、腎臟結構改變等,如果不能及早診斷,可能會導致進展性慢性肝病或肝功能衰竭,甚至死亡。在肝硬化及肝衰竭前診斷此病,給予口服初級膽汁酸替代治療,可使臨床癥狀顯著改善甚至臨床治愈。分子遺傳學檢測是目前診斷該病的金標準。在臨床工作中若高度懷疑此病,應盡早行基因檢測以明確診斷。進一步探索HSD3B7基因突變的基因型與表型之間的關系,可對指導個體化治療、預后的判定和遺傳咨詢具有重要意義。
